Synopsis
Consistent with a protective role, under static oxic conditions we found that FeII oxidation in the presence of added 13C-DOM resulted in SRO Fe–C associations that not only inhibited the mineralization of 13C-DOM by 35% relative to controls, but also suppressed the priming of native SOM mineralization by 47%, which consequently decreased overall CO2 production by 22% (Fig. 1d). However, when 13C-DOM was not added, FeII oxidation and the production of reactive oxygen species stimulated mineralization of native SOM by 8% relative to the controls (Fig. 1d). Thus, the formation of additional SRO–Fe phases did not provide net protection to SOM unless there was additional DOM present. As might be expected, the protective role of Fe was reversible under anoxic conditions. Although CO2 production from non-Fe amended treatments during the anoxic period was 68–70% lower than in the static oxic treatment (Fig. 1d), the de novo SRO Fe–MAOM formed via FeII oxidation was disproportionately vulnerable to subsequent reduction. This consequently stimulated the mineralization of both added 13C-DOM and the native SOM by 74% and 32–41%, respectively, and thus increased overall CO2 production by 41–49% relative to both non-Fe amended treatments (with or without added DOM, Fig. 1d). As a result of Fe-stimulated C mineralization, the anaerobic 13C-DOM mineralization was 81% greater than the oxic control. Below we provide details on the production of the Fe–MAOM, discuss the data supporting Fe protection of C along with the data supporting Fe stimulation of C loss, and then provide a synthesis of the work.
Generation of FeIII-(oxyhydr)oxides
The oxidation of 57FeII after a 1-d equilibration with the soil under anoxic conditions generated SRO FeIII (oxyhydr)oxides that impacted C cycling. Exposure to O2 (day 1–6) led to the oxidation of FeII, with aqueous FeII completely oxidized within 6 h. The sorbed FeII substantially decreased by 91% over the first day and slowly declined thereafter (Fig. 2). The treatment with both 57Fe and 13C-DOM added had 10% more adsorbed 57FeII than the 57FeII-only treatment before oxidation (Fig. 2a and b), likely due to co-sorption of the Fe2+–DOM complex, as observed previously44. The variable-temperature Mössbauer spectroscopy technique that we use to track the mineral composition of the 57Fe additions, gives excellent information on the crystallinity of the Fe phases, with high crystallinity phases ordering at higher temperatures. Both 57Fe addition treatments led to the formation of de novo SRO 57FeIII phases of lower crystallinity (lower Mössbauer ordering temperature) than the bulk soil Fe (Table 1; Supplementary Fig. 2 and Fig. 3), resulting in a 26–31 mmol kg−1 increase in lepidocrocite and 3–14 mmol kg−1 increase in nanogoethite and very-disordered FeIII (oxyhydr)oxides that preclude assignment (Fig. 3; Supplementary Table 1). The addition of 57Fe and 13C-DOM together resulted in the formation of even lower crystallinity SRO FeIII (oxyhydr)oxides than the 57Fe addition-only treatment as illustrated by the lower 35K/5K and 12K/5K crystallinity ratios (Table 1; Supplementary Fig. 3). Suppression of FeIII crystallinity by co-precipitation with dissolved fulvic acids has been shown previously in synthetic pure systems44, and here we extended this finding to a complex soil system containing a mixture of aluminosilicates, FeIII (oxyhydr)oxides, and a variety of organic compounds. In general, lower crystallinity Fe (oxyhydr)oxides (often measured by oxalate-extraction) have higher surface area, sorb more OM, and are thought to be associated with persistent OM in soils45.
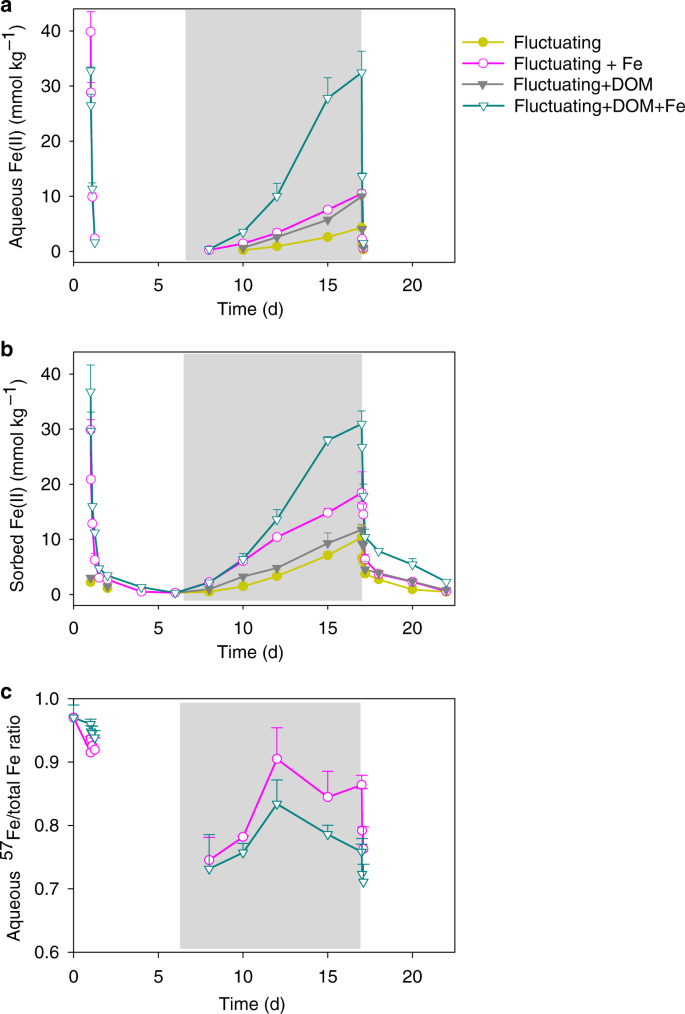
Time-dependent a aqueous FeII, b sorbed (HCl-extractable) FeII, and c dissolved 57Fe to total Fe ratio in the aqueous phase in the redox-fluctuating treatment. FeII and 57Fe were undetectable during day 1.5–6 and 17.5–22. The gray shaded region represents the anoxic phase of the redox-fluctuating treatment. Error bars indicate s.e.m. (n = 3).
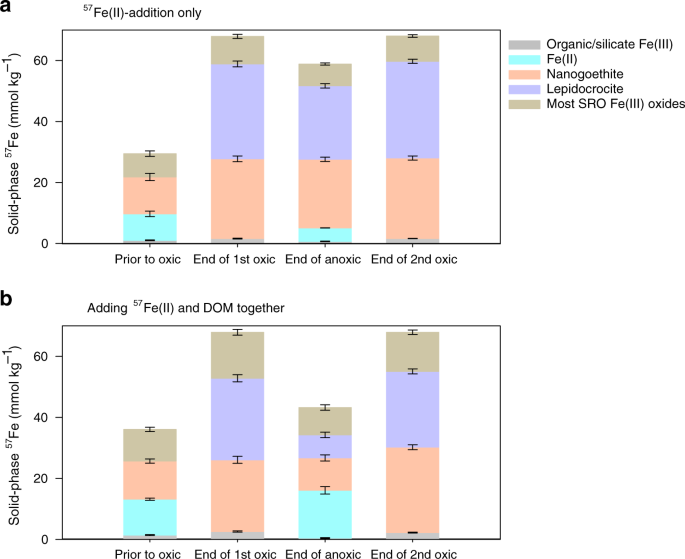
57Fe partition was calculated from respective Mössbauer spectra (corrected to exclude the signal from the native soil Fe) for the (a) 57Fe-only and (b) 13C-DOM-57Fe addition treatments, prior to the oxic phase (day 1) and at the end of the 1st oxic (day 6), anoxic (day 17) and 2nd oxic (day 22) phases. Error bars represent standard errors associated with Mössbauer data modeling (see Supplementary Information).
Iron protection of organic matter
In the static oxic treatment, addition of 57Fe suppressed the mineralization of 13C-DOM by 35% (p < 0.01, Figs. 1d, 4 and Table 2): cumulative CO2 production was 25.6 ± 0.8 and 39.5 ± 1.1 mmol C kg−1 with and without 57Fe, respectively, equivalent to 17.1% and 26.3% of the added 13C-DOM (Fig. 4c and Table 2). Although 57Fe addition also inhibited net 13C-DOM mineralization in the fluctuating redox treatments (p < 0.01, Fig. 4c and Table 2), this inhibition was confined to the oxic portions (days 1–6 and days 17–22) of the incubation and was partly offset by a 74% enhanced 13C-DOM mineralization during the anoxic phase (days 6–17) relative to the treatment without added Fe (Figs. 1d, 4b, and Table 2, see below).
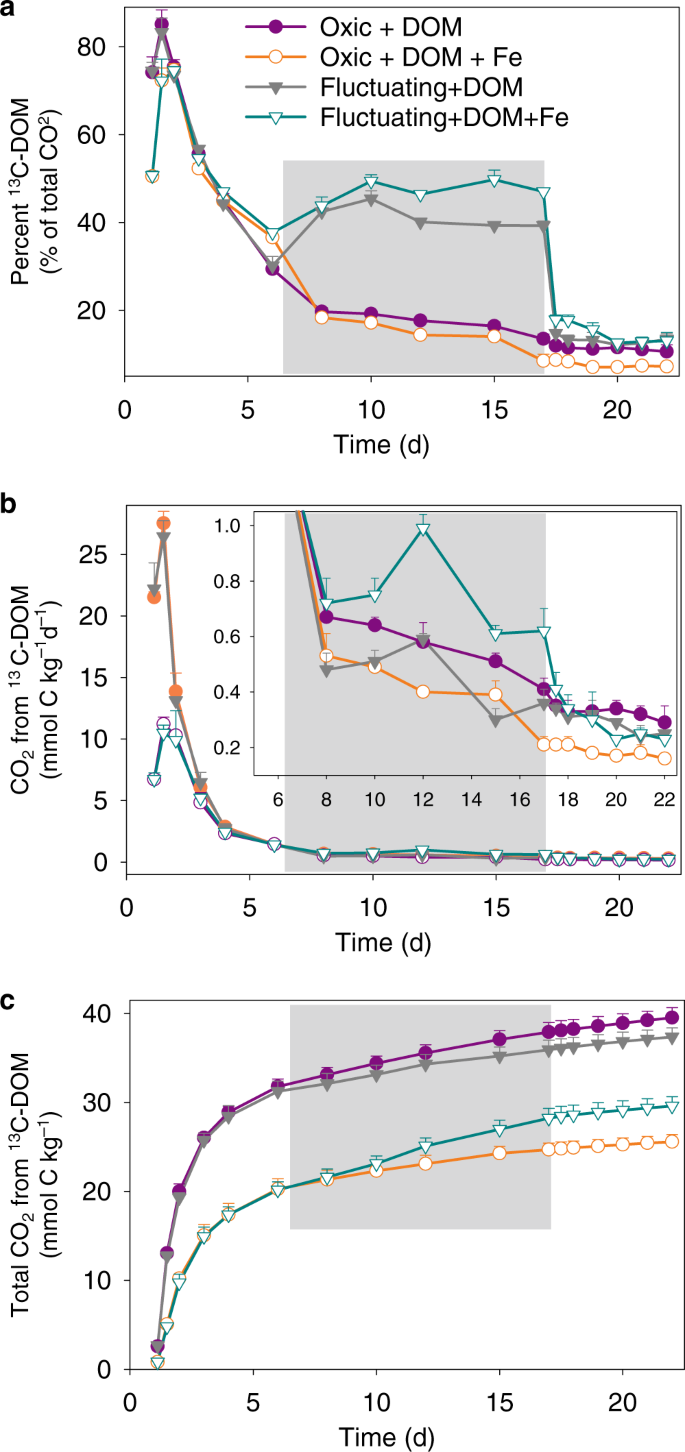
a The percent contribution of 13C-labeled DOM to total CO2 production, b CO2 production rates from 13C-labeled DOM, and c cumulative CO2 production from 13C-labeled DOM. The gray shaded region represents the anoxic phase of the redox-fluctuating treatment. Error bars indicate s.e.m. (n = 3).
The generation of low crystallinity SRO–FeIII (oxyhydr)oxides from the oxidation of 57FeII in the presence of 13C-DOM resulted in a lower DOC concentration than in the 13C-DOM-only treatment and a concurrent increase in solid-phase 13C content (Fig. 5). This likely reflects the formation of SRO Fe–C complexes with 13C-DOM adsorbing or co-precipitating with the newly-formed SRO lepidocrocite and nanogoethite phases (Fig. 3). It is generally assumed that SRO Fe phases contribute to soil C persistence by protecting it against microbial mineralization5, but few studies have directly measured the bioavailability of Fe-associated OM39. Our study provides evidence that de novo formation of SRO Fe–C complexes inhibit the mineralization of fresh DOM inputs to soil. Others have also observed a large decrease in OM decomposition when glucose or fulvic acid sorbed to synthetic Fe minerals (ferrihydrite/goethite) was added to soils, as compared to additions of the free organic compounds40,41. The bioavailability of mineral-associated OM is generally thought to be linked to C loadings (e.g., C/Fe ratios), with a maximum adsorption capacity occuring at a C/Fe molar ratio of about one46. Co-precipitation could result in Fe–OM associations with much higher C/Fe ratios11,12,19. In our study, the initial C/Fe molar ratio of the added 13C-DOM and 57Fe was 2.1. If we assume that all DOM that was removed from the solution during the FeII oxidation event sorbed to the newly-formed FeIII (oxyhydr)oxides, the C/Fe ratio of those OM–FeIII (oxyhydr)oxide complexes would be ~1.7. Thus, there was likely 13C-DOM with a low affinity for FeIII (oxyhydr)oxides that remained as unprotected 13C-DOM in the aqueous phase and this likely led to our observation of significant 13C-DOM mineralization even in the presence of de novo FeIII (oxyhydr)oxides (Fig. 4).
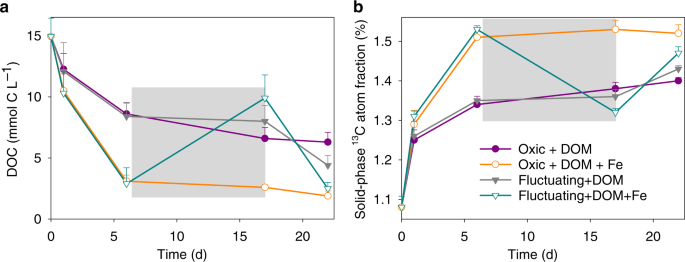
a Dissolved organic carbon concentration (MES buffer concentration was subtracted) and b solid-phase 13C atom fraction from 13C-DOM addition treatments. The gray shaded region represents the anoxic phase of the redox-fluctuating treatment. Error bars represent s.e.m. (n = 3).
Labile C inputs are often observed to alter the decomposition of extant SOM, defined as priming47,48. During the oxic periods of the experiment, 57FeII oxidation in the presence of added 13C-DOM not only suppressed the mineralization of the amended 13C-DOM, but also partially inhibited the priming of native SOM decomposition compared to the DOM-only treatment (Fig. 6; Table 2; Supplementary Table 2). In the static oxic treatment, addition of 13C-DOM alone or together with 57Fe increased native SOM-derived CO2 production compared to the soil-only control (priming effect) (p < 0.01, Fig. 6a, c and e; Table 2; Supplementary Table 2). However, adding 13C-DOM and 57Fe together resulted in a significantly smaller priming effect on native SOM mineralization than adding 13C-DOM alone under the static oxic treatment (p < 0.01, Fig. 6e, Table 2 and Supplementary Table 2). With the addition of 13C-DOM, cumulative primed CO2 from native SOM under the static oxic treatment measured 10.2 ± 1.2 and 19.1 ± 0.9 mmol C kg−1 with and without 57Fe addition, respectively (Fig. 6e and Supplementary Table 2). Cumulatively, adding 13C-DOM together with 57Fe suppressed aerobic priming of native SOM by 47% relative to adding 13C-DOM alone (Fig. 1d, Table 2 and Supplementary Table 2). Collectively, 57Fe oxidation in the presence of 13C-DOM resulted in 22% less overall C mineralization, compared to addition of 13C-DOM alone (Fig. 1d and Table 2).
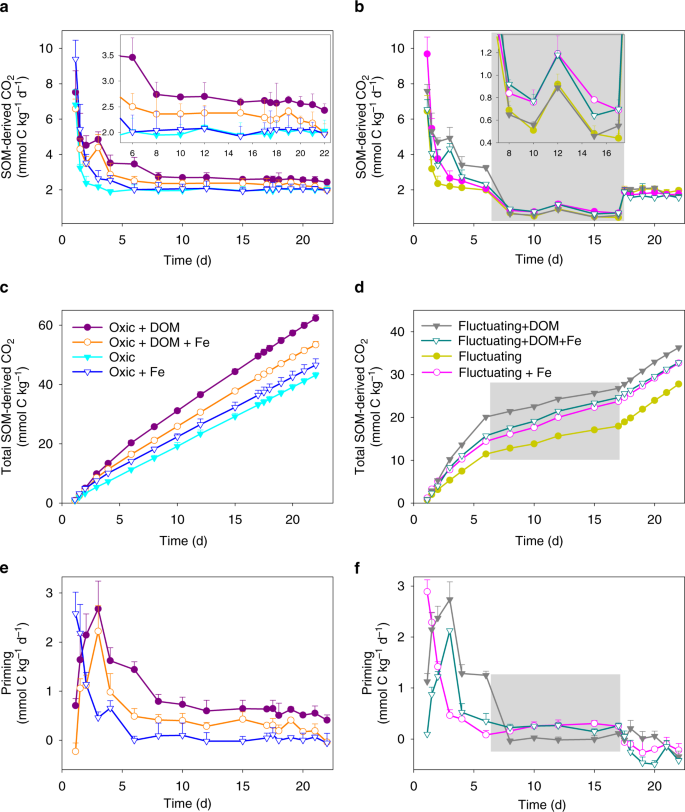
CO2 production rates from soil C under a static-oxic and b redox-fluctuating conditions, cumulative CO2 production from soil C under c static-oxic and d redox-fluctuating conditions, and priming of soil C under e static-oxic and f redox-fluctuating conditions. The gray shaded region represents the anoxic phase of the redox-fluctuating treatment. Error bars indicate s.e.m. (n = 3).
We propose that de novo SRO FeIII minerals protected the added 13C-DOM under static oxic conditions, decreasing DOM availability for microbial growth, and suppressing the priming of native SOM. An alternative explanation, that the sorption of native SOM onto the de novo SRO FeIII (oxyhydr)oxides inhibited priming, is unlikely because when we added 57FeII alone it actually increased the mineralization of native SOM (due to reactive oxygen species production, as discussed below) (Fig. 6a, e and Table 2). Similarly, prior studies have shown that the addition of new SRO FeIII phases to soils has little to no impact on the mineralization of native SOM41,49. Rather, it is likely that DOM–FeIII interactions create physico-chemical barriers that limit priming by decreasing microbial access to the new DOM. Thus, when reduced soils receive oxygenated water due to rainfall, snowmelt, or irrigation, the oxidation of FeII in the presence of DOM and formation of OM–FeIII complexes may contribute to C protection both directly, as previously known, and indirectly, by suppressing priming.
Iron stimulation of DOM and SOM mineralization
Only in the treatment where 13C-DOM and 57Fe were added together were we able to confirm that Fe had an overall protective effect on OM, and that protection was limited to the oxic portions of the experiment. Below, we quantified the impact of Fe on OM mineralization via Fe-stimulated Fenton chemistry during the first few days of oxic exposure and via Fe reduction-mediated reactions during the anoxic periods.
Adding 57Fe alone strongly stimulated CO2 production from native SOM during the first 3 days of the static oxic treatment (and the oxic portions of the fluctuating redox treatment) relative to the soil-only control (p < 0.01), with no stimulatory impact afterwards (Fig. 6a and e). Cumulatively, FeII oxidation stimulated CO2 production by 8% (Fig. 1d and Table 2). To confirm the role of Fenton chemistry, we performed a parallel experiment with added terephthalate—an effective hydroxyl radical scavenger—and found similar CO2 production between FeII-added treatments and soil-only controls (Supplementary Fig. 4). Recent studies have similarly shown that FeII oxidation is linked to increases in soil CO2 production via the generation of radical oxygen species50,51, which facilitate the breakdown of complex biopolymers to produce labile substrates for microbial respiration34,35. Others have also attributed increased CO2 production following FeII oxidation to an increase in acidity that can promote DOC release36. Given that we conducted these experiments in a strong buffer at a constant pH, the increased CO2 production following FeII oxidation was most likely derived from the production of reactive oxygen species such as the hydroxyl radical.
Soil C mineralization rates typically decrease as O2 becomes limiting22,52,53. In our soil-only control, CO2 production from native SOM during the anoxic period was 70% lower than in the static oxic treatment (Figs. 1d, 6 and Table 2). However, during the anoxic portions of the experiment, Fe addition stimulated native SOM mineralization relative to the no-Fe treatment (Fig. 6b and f; Table 2). In the 57Fe-addition treatment, the degree of anoxic suppression of CO2 production decreased from 70 to 58% of that under oxic conditions (p < 0.05; Table 2), as a result of a 41% higher anoxic native SOM-derived CO2 production in the Fe addition treatment than in the no-addition control (p < 0.05, Table 2; Figs. 1d, 6b, d and f). This likely resulted from enhanced microbial use of FeIII as an electron acceptor in the 57Fe addition treatments. Following the transition from oxic to anoxic conditions in the fluctuating redox treatments, substantial FeIII reduction occurred (day 6–17, Fig. 2a and b) and adding 57Fe increased the total FeII production rates (2.9 mmol kg−1 d−1) compared to the soil-only control (1.6 mmol kg−1 d−1). This was most likely due to the facile reduction of de novo 57Fe SRO lepidocrocite and nanogoethite, which had a much lower crystallinity (and thus a higher reactivity) than native soil FeIII (oxyhydr)oxides (Table 1 and Fig. 3; Supplementary Table 1 and Fig. 3). The availability of native SRO FeIII phases likely limits Fe reduction in this subtropical agricultural soil, and the de novo SRO 57FeIII phases were preferentially utilized as electron acceptors for microbial respiration as evidenced by the preferential release of 57FeII in the aqueous phase (Fig. 2c) and the measured decrease of these 57Fe mineral phases following reduction (Fig. 3a and Table 1; Supplementary Table 1).
Iron’s stimulation of C mineralization during anoxic periods was greatly enhanced when 57Fe and 13C-DOM were added together, yielding increases in mineralization of native SOM (anoxic priming) and 13C-DOM by 32 ± 3% and 74 ± 7%, respectively, relative to adding 13C-DOM alone (p < 0.05; Table 2; Figs. 1d, 4b, 4c, 6b, d and f). In fact, when13C-DOM and 57Fe were added together, anaerobic 13C-DOM mineralization in the fluctuating redox treatment was even 81% greater than the aerobic 13C-DOM mineralization in the static oxic treatment at the same point in time (p < 0.01; Fig. 4b; Table 2). The stimulation of 13C-DOM mineralization under anoxic conditions was linked in part to its molecular composition, given that thermodynamic constraints on Fe reduction limit metabolism to relatively oxidized C substrates22,42,43. During the anoxic periods, the mineralization of 13C-DOM over the native SOM (in both the 13C-DOM only and DOM–Fe addition treatments) was 2–3 times higher than that in the static oxic treatment at the same point (Fig. 4a). Characterization of the molecular composition of 13C-DOM and water-extractable native SOM using Fourier transform ion cyclotron resonance mass spectrometry (FTICR-MS) revealed that the 13C-DOM had significantly less lignin-derived materials and much more aliphatic formulae than the water-extractable native SOM (Supplementary Table 3, Figs. 5 and 6), which represents the most bioavailable fraction of native SOM54. The preferential anaerobic mineralization of 13C-DOM over SOM may be due to a lower abundance of lignin-derived compounds, which are not readily depolymerized under anoxic conditions55. In addition, compared to water-extractable native SOM, 13C-DOM contains compounds with higher nominal oxidation state of C (NOSC values > 0.5, Supplementary Fig. 6), which are associated with a higher likelihood of thermodynamic favorability (−ΔGr) when coupled to FeIII reduction than the bioavailable fraction of native SOM22,43. Fe reduction was also stimulated by the addition of 13C-DOM alone (producing 2.3 mmol kg−1 d−1 of FeII compared to the soil-only control rate of 1.6 mmol kg−1 d−1) (Fig. 2a and b), consistent with prior work26. However, when Fe and DOM were added together, Fe reduction was greatly increased to 7.4 mmol kg−1 d−1, which was even greater than the additive effect of separate 13C-DOM (2.3 mmol kg−1 d−1) and 57Fe additions (2.9 mmol kg−1 d−1) (Fig. 2a and b). This was because compared to oxidation of 57FeII alone, oxidizing 57FeII in the presence of 13C-DOM led to formation of even less-crystalline SRO lepidocrocite and nanogoethite phases (all ordering at <35 K in the Mössbauer spectra, Table 1 and Supplementary Fig. 3). These SRO 57Fe-13C-OM phases exhibited high rates of Fe reduction, releasing significant 57Fe2+(aq) and 13C-DOM (Figs. 2 and 5) when exposed to anoxic conditions, leaving the solid phase depleted in its lowest crystallinity Fe phases (Fig. 3 and Table 1; Supplementary Table 1 and Fig. 7), and preferentially stimulating anaerobic mineralization of the added 13C-DOM (Fig. 4a).
Fe reduction can solubilize significant amounts of OM adsorbed or coprecipitated with FeIII (oxyhydr)oxides directly, as shown in our experiment, or indirectly because of an increase in pH30,32,56. This re-mobilized 13C-DOM often includes biochemically labile C32,57, and may potentially offset the kinetic/thermodynamic constraints often limiting anaerobic decomposition22,43,58. We find that collectively, the reduction of SRO FeIII phases offset O2 limitations on C mineralization by 24 ± 3% relative to the non-Fe amended treatment (Table 2).
Synthesis
A recent survey of over 5500 soil profiles spanning continental scale environmental gradients found that SRO Fe and Al (oxyhydr)oxide abundance was the best predictor of C content in humid soils, among the geochemical and climate variables that were available45. This is consistent with other work showing that SRO FeIII phases are broadly implicated in the persistence of OM in soil1,3,59. However, the nature of the relationship between Fe and C in humid soils—and redox dynamic soils in general, which would include floodplain and perennial wetland soils from all climatic regions—is far from straightforward. Humid soils are replete with microsites that undergo dynamic anoxia in response to high labile C loads during periods of high moisture and experience appreciable FeIII reduction rates23,25,60,61. Oxidation of the FeII generated from FeIII reduction is a common mechanism for MAOM formation in humid and redox-dynamic soils, yet Fe is also responsible for OM loss and our work here illustrates two principal refinements in this regard.
First, the production of SRO Fe–MAOM via FeII oxidation will likely increase CO2 production in the short-term. Only when we formed MAOM in the presence of DOM and maintained strict oxic conditions was there a net decrease in C mineralization (both in the added 13C-DOM and the native SOM, i.e. via decreased priming). When we simply generated MAOM via FeII oxidation without added DOM, Fenton chemistry caused an 8% increase in C mineralization (Fig. 1d). Upon the inevitable return to periodic anoxia in humid soils, our work suggests that C mineralization would be accelerated by 41–49% by Fe reduction (Fig. 1d), thus counteracting the stabilization effect on OM of SRO Fe phases. The magnitude of these counteracting mechanisms may also be influenced by soil structure, which we largely eliminated in our study by conducting experiments in soil slurries. Hence, direct application of our results to in situ soil environments is tentative. However, the general principles of our work are also likely to be applicable to structurally complex soil systems. For example, Fe mineral-associated C is often released in natural soils under in-situ flow conditions as a consequence of dissimilatory Fe reduction (e.g.,62) and thus becomes more vulnerable to microbial decomposition. In our study, we even found that the added DOM was preferentially degraded under anoxic conditions relative to the oxic control (Fig. 4), which highlights how the thermodynamic constraints of anaerobic metabolism and the molecular composition of C sources can influence the fate of fresh DOM inputs22,42,43. Consequently, the net effect of Fe–C interactions in dynamic redox environments likely hinges in part on the composition of DOM inputs, a worthy topic for further research.
Second, our work here suggests that the initial SRO Fe–C associations are not likely to persist without protection from periodic Fe reduction events. Several researchers have identified or produced SRO–FeIII–OM colloids that are resistant to either microbial or chemical reduction63,64,65,66,67, however, the key components conferring this protection are variable and/or elusive. Some work has identified that SRO–FeIII–OM co-precipitates with low C/Fe ratios provide resistance to microbial reduction63,64, whereas other work has emphasized structural properties (conformation and micro-aggregation) as the mechanism that retards dissolution65,66,67. SRO Fe–OM phases are often co-precipitated with Al and Si ions68—which can retard recrystallization69—and given the co-association of Al and Fe with OM in humid soils, Al is a strong candidate for protecting Fe against reduction. However, studies that have examined Al and Si co-precipitated Fe-(oxyhydr)oxides found those ions also make the co-precipitates more susceptible to reductive dissolution70. Coward et al.67 recently proposed several mechanisms by which SRO FeIII–OM phases could become resistant to reductive dissolution, including acquiring reduction-resistant surface coatings, or becoming embedded in a composite aggregate structure6. Such a protective coating could even come from higher crystallinity Fe (oxyhydr)oxides. Hall et al. recently found that 14C-derived C residence time in humid soils was positively correlated with Fe phase crystallinity71. Consistent with that, we find here that in contrast to the initial oxidation event, the 2nd oxidation event generated more crystalline 57Fe phases (Table 1; Supplementary Fig. 8) and did not stimulate additional C mineralization (Fig. 6 and Table 2). It may be that during repeated redox fluctuations a substantial portion of the co-precipitated OM would be lost, but a core Fe–MAOM structure would remain protected from reductive dissolution.
Perhaps most compelling is the growing evidence that various aggregation, conformation, and structural characteristics of soils confer protection for OM5,6,7,10. Even the protective surface coatings66,67 or conformational changes in OM at low C/Fe ratios64 discussed above are examples of micro-aggregate structures not unlike the encasement of SRO Fe–OM phases by aluminosilicate clays or other processes that generate micro-aggregates of minerals and OM during pedogenesis6,7,10,72. These aggregation processes can structure microaggregates with core SRO Fe phases and outer aluminosilicate or other phases that are not susceptible to reductive dissolution—as observed in Andisols by dithionite-resistant SRO–Fe phases66. Our soil slurry approach was designed to minimize the physical constraints (macro-pore flow, spatial arrangement of microbes, minerals and OM, and the development of aggregates) on C decomposition and thereby isolate the sorbent and electron-transfer roles of Fe in C dynamics (Supplementary Fig. 1). Under these conditions, we find that Fe does not confer intrinsic protection for OM in redox-dynamic soils. In an in situ soil environment—where MAOM emerges in a dynamic three-dimensional space—structural and physical protection of MAOM is thus likely a key protective mechanism for reconciling the comparatively large proportions of SRO-OM associations in soil of very old age based on 14C-dating1,4,5,59. Future studies should thus assess the extent that the formation and destruction of Fe-cemented microaggregates contribute to OM persistence in redox-dynamic soils. Our work demonstrates that the inherent persistence of SRO Fe-associated C cannot be guaranteed. Biological and geochemical context is critical for understanding the long-term fate of FeIII-associated SOM under a changing climate, given the dual roles of FeIII phases in both accelerating and inhibiting OM decomposition.
Source: Ecology - nature.com
