General features of collected samples and characteristics of sequenced data
We analysed a total 16 adult samples (NWP1 n = 8; NWP2 n = 4; NWP3 n = 4) and 12 juvenile samples (NWP1 n = 6; NWP2 n = 6) from three cryptic species (NWP1, NWP2, and NWP3) in the grey mullet M. cephalus species complex in the Taiwan Strait during their spawning season. Fish samples were identified as specific cryptic species based on methods from previous studies15,16 (see Materials and Methods). The diversity and structure of the gut microbial composition were determined by performing deep sequencing of bacterial 16S rRNA genes from all fish gut samples. After quality filtering and removing chimeras and single reads, a total of 3,704,851 high-quality reads were obtained from 16 adult (hereafter AD) and 12 juvenile (JV) grey mullet samples. The number of reads ranged from 75,613 to 200,144 reads of 16S rRNA amplicons in each sample (Supplementary Table 1), resulting in the identification of 1,160 operational taxonomic units (OTUs).
Evaluation of the microbial complexity in the M. cephalus species complex
In this study, 750,613 reads were rarefied in all samples. Rarefaction curves approached the saturation plateau and adequately represented the gut microbial community diversity in all samples (Supplementary Fig. 1). Although there was no difference between the results of the analyses using rarefied and unrarefied data, it is believed that rarefied data can ignore the presence of rare species, which leads to false positives17. Therefore, we prioritized the results from the unrarefied data. At the species level, wild AD grey mullets had an average of 123 ± 9, 177 ± 39, and 166 ± 57 OTUs in NWP1, NWP2, and NWP3, respectively. Meanwhile, we detected 566 ± 64 OTUs in NWP1 and 489 ± 95 OTUs in NWP2 juveniles. Inter-specific variation among individuals in sampled species were examined using a permutational test for homogeneity of multivariate dispersions. Although the box plot showed higher inter-variation between individual samples of NWP3 juvenile adult (Supplementary Fig. 2), the p value was not significant (p = 0.292). The alpha diversity—including observed richness (sob), Chao diversity, Shannon index (H’) and inverse Simpson diversity (S) indices—were calculated. In addition, permutation T-test was conducted to test whether there was any difference in alpha diversity among adult groups, among juvenile groups, and between adults and juveniles of the same cryptic species. The results showed that, while there are similarities between the alpha diversities of AD_NWP1 and AD_NWP3 and between JV_NWP1 and JV_NWP2 in both indices, there was significantly more diversity in juvenile samples compared to adults of the same cryptic species (AD_NWP1 – JV_NWP1: Psob < 0.01, Pchao < 0.01, PH’ < 0.01, PS < 0.01; AD_NWP2 – JV_NWP2: Psob < 0.05, Pchao < 0.05, PS < 0.05). At the same time, AD_NWP2 had the lowest alpha diversity (except in the Chao estimation) compared to AD_NWP1 in the observed richness and Shannon and Inverse Simpson indices and AD_NWP3 in the Inverse Simpson index (Fig. 2, Supplementary Table 2).
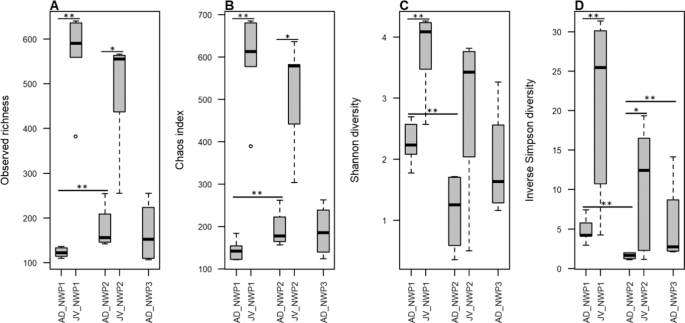
Alpha diversity in the grey mullet gut microbiota. (A) Observed richness, (B) Chaos diversity index, (C) Shannon diversity, and (D) inverse Simpson diversity indexes were calculated following rarefaction-based normalization of the OTU table. Significant p values (*p < 0.05; **p < 0.01) were obtained using a permutation T-test. X axis shows the group IDs; JV is juvenile, AD is adult, NWP1 is cryptic species 1, NWP2 is cryptic species 2, and NWP3 is cryptic species 3.
Gut microbial composition
The structure of the gastrointestinal microbiota was characterized using the relative abundance of representative sequences from each OTU from each sample to assign a taxonomic classification against the Greengenes database. A total of 45 different cultured and candidate bacterial phyla were detected from all fish gut samples. In adult samples, M. cephalus gut microbiota across the phylum were dominated by Proteobacteria (65.4–89.5%) in cryptic species 1 (NWP1) while the most abundant phylum of bacteria was Actinobacteria (74–94.5%) in cryptic species 2 (NWP2) and Spirochaetes (14.8–66.6%) in cryptic species 3 (NWP3). Furthermore, the gut microbiota of juvenile samples NWP1 were dominated by Proteobacteria (37.9%), Cyanobacteria (33.9%), and Firmicutes (12.5%), while the NWP2 juvenile gut microbial community consisted of high abundances of Firmicutes (46.4%), Proteobacteria (32.3%), and Actinobacteria (13.7%) (Fig. 3A).
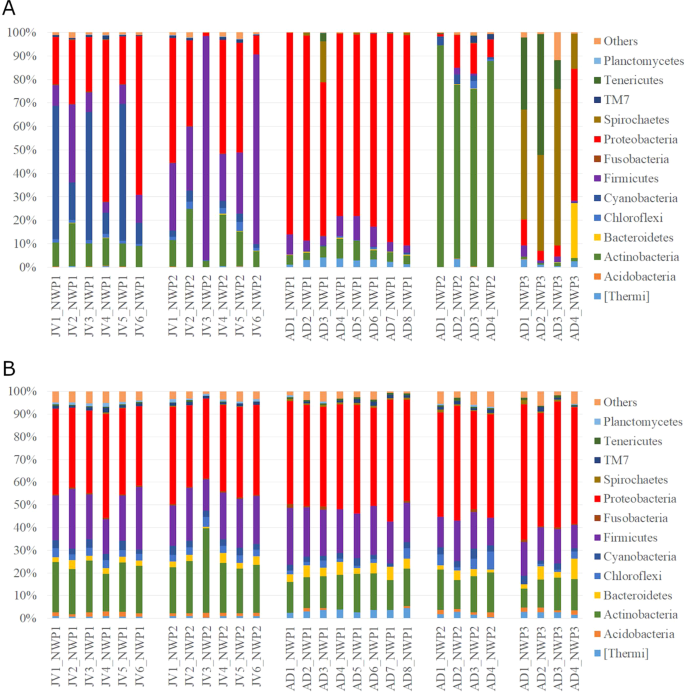
The gut microbial communities in grey mullets at the phylum level. (A) The structure of gut microbial compositions (relative abundance of phyla). (B) The membership of gut microbial communities (number of taxa present in each phylum category). Others categories include unclassified bacteria and low abundant phyla. X axis shows the sample IDs; JV is juvenile, AD is adult, NWP1 is cryptic species 1, NWP2 is cryptic species 2, and NWP3 is cryptic species 3. There were 12 juvenile samples (NWP1 n = 6; NWP2 n = 6) and 16 adult samples (NWP1 n = 8; NWP2 n = 4; NWP3 n = 4).
At the family level, Moraxellaceae was the most abundant bacterial group in NWP1 adults (accounting from 43.7–77.7%), while one family in the order Actinomycetales dominated 70.2–98.9% of the gut microbial community in NWP2 adults. Samples belonging to cryptic species NWP3 had high abundances of Brevinemataceae and Mycoplasmataceae bacteria (14.9–92.3%). In juvenile samples, Streptococcaceae, Clostriaceae, Vibrionaceae, and one unclassified family belonging to order Stramenopiles were the most representative of the gut microbial compositions, contributing to 30–94.5% of the total abundance (Supplementary Fig. 3).
In contrast to the relative abundances of gut bacteria, our analysis detected similar proportions of community membership among all grey mullet fish, including adult and juvenile samples. Community membership is calculated as the number of bacteria taxa at the phylum level. Specifically, Proteobacteria, Firmicutes and Actinobacteria accounted for an average of 44, 19, and 18% of identified OTUs in all fish samples, respectively (Fig. 3B).
We next used beta diversity analysis to approximate the variation in gut microbial community structure inside the M. cephalus species complex. Non-metric multidimensional scaling (NMDS) plots showed that grey mullet gut microbial compositions were distinctively separate from different cryptic species in adult samples, and were strongly clustered between juvenile fishes based on Bray Curtis and weighted UniFrac distance matrixes (Fig. 4). PERMANOVA analyses also supported our delineated results (Supplementary Table 3). In particular, there were differences in microbial composition among adults of the three cryptic species (p < 0.05). The microbial communities in adults and juveniles of the same cryptic species were also significantly different (p < 0.001). On the other hand, there were no species-specific differences observed between juvenile samples of NWP1 and NWP2.
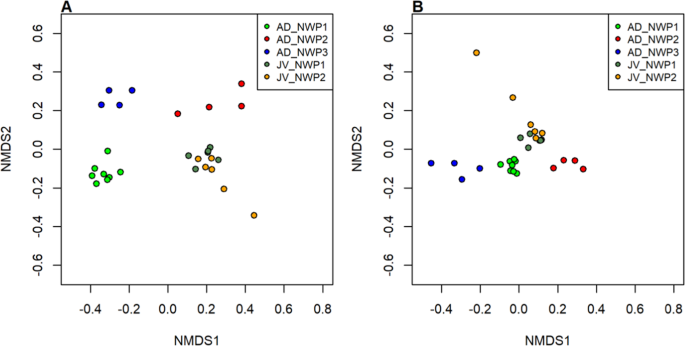
Two-dimensional non-metric multidimensional scaling (NMDS) plots of gut microbial communities. The gut microbiota compositions of 16 adult and 12 juvenile grey mullet samples were used to estimate the similarity between samples based on (A). Bray Curtis distance, and (B). weighted UniFrac distance. Light green points are gut microbial communities in NWP1 adult samples, red points for communities in NWP2 adults, blue points for communities in NWP3 adults, and dark green and orange dots are juvenile samples of NWP1 and NWP2, respectively. NWP1, NWP2 and NWP3 are three cryptic species of grey mullet M. cephalus in the Taiwan Strait.
Core gut microbiome of M. cephalus
To investigate the existence of a core gut microbiome that is maintained across the M. cephalus life history, we focused on 122 genus-level OTUs shared in all the fish groups in this study. Consistent with membership of gut microbiota in grey mullets, most of the core microbes were Proteobacteria (56 OTUs, 45.9%), Firmicutes (22 OTUs, 18.03%), and Actinobacteria (18 OTUs, 14.75%). The fact that the relative abundance of the core gut microbes can be as high as 95.8% in the communities might reflect the importance of bacteria inside the gut microbial community, as well as their potential functions in and benefits to the hosts. Within these 122 OTUs, 35 were present in over 90% of observed samples. Permutation T-test results indicated that many core microbes inside the gut microbial community in AD_NWP1 were significantly different with not only the other adult cryptic species (NWP2 and NWP3; 22 and 13 OTUs, respectively), but also the juveniles of this cryptic species (31 OTUs). OTU499 (assigned to a genus of Brevinemataceae) was the only OTU with a notably different abundance among the gut microbiota of all three cryptic species in the adult stage (PNWP1–NWP2 < 0.05, PNWP1–NWP3 = 0.01, PNWP2–NWP3 < 0.05) (Table 1).
Clustering of juvenile gut microbial composition
From the 827 OTUs detected at the genus level in the gut microbial communities of 12 juvenile grey mullets (NWP1 n = 6 and NWP2 n = 6), we identified 598 OTUs shared between the two cryptic species, which accounted for 99.75% and 99.87% of the relative abundance in the gut microbial communities of NWP1 and NWP2 juveniles, respectively (Fig. 5A; Supplementary Table 3). High similarities in libraries between juveniles is consistent with the results of the NMDS plots from the Bray-Curtis distant and weighted UniFrac (Fig. 4). In particular, juvenile samples’ gut microbial communities clustered together, and the PERMANOVA test found that they were not significantly different. T-test results showed that only 17 of the 598 shared OTUs were significantly different between NWP1 and NWP2 juveniles (Fig. 5B).
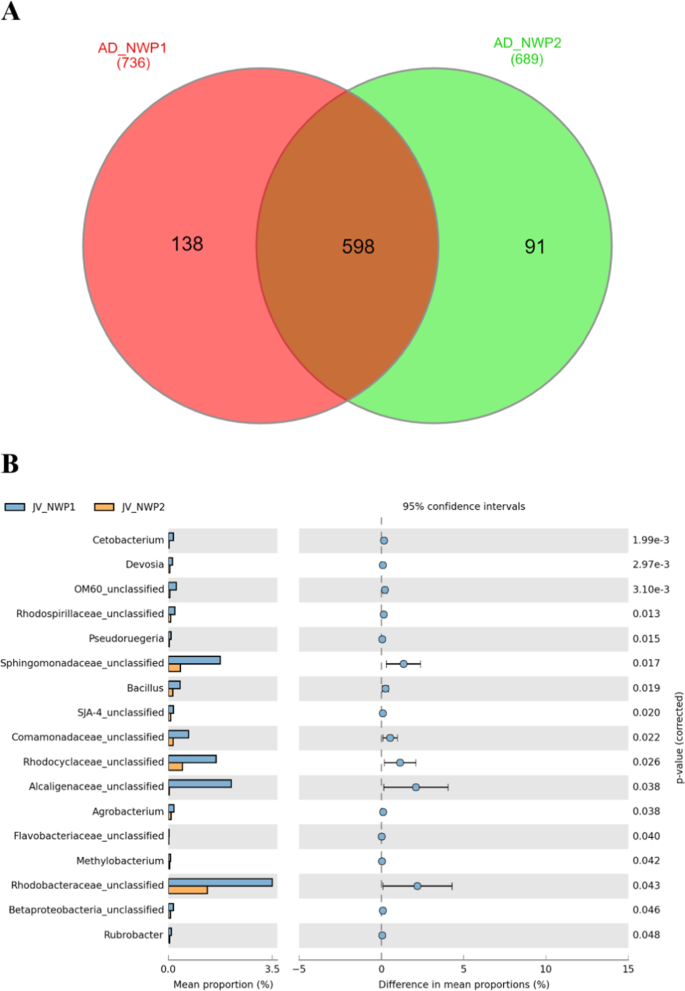
Analysis of the shared OTUs in grey mullet juveniles. (A) Venn diagram indicates the unique and shared OTUs between NWP1 juveniles (red orange) and NWP2 juveniles (green). (B). Significant different shared OTUs between juveniles of NWP1 (blue) and NWP2 (orange). p values were calculated using T-test, and p value < 0.05 was considered statistically significant. NWP1 and NWP2 are two cryptic species of grey mullet M. cephalus in the Taiwan Strait.
Cryptic species-specific microbiome
In addition to the core microbiome at the species complex level, we clarified unique shared OTUs in specific cryptic species. Fourteen OTUs were only observed in NWP1, while six were only found to be shared between adults and juveniles of NWP2. We ran sequences of unique and shared OTUs in two cryptic species through NCBI BLAST, which identified species names with NCBI IDs. Next, we generated phylograms between identified OTUs using Maximum Likelihood analysis based on the Kimura 2-parameter model (Supplementary Fig. 4). Interestingly, even though its gut was dominated by Actinobacteria in the adult stage, four of the six unique OTUs in NWP2 were Proteobacteria (i.e. Neisseria sp., Aquabacterium sp., Rheinheimera sp.and Chujaibacter sp.). At the same time, three Actinobacteria species (Corynebacterium sp., Agrococcus sp. and Janibacter sp.) were only detected in NWP1. Moreover, we detected two closely related Neisseria spp. in the gut microbiota of these two different cryptic species.
Potential impact of seawater microflora on the fish gut microbial community
The influence of host genetics on the gut microbiota was partly reflected in the similarities in communities among juvenile fishes of two closely-related cryptic species, as well as the observation that compositions harboured almost identical membership in all the fish gut samples. However, the clear distinction between structures of gut microbial compositions in adults belonging to three different cryptic species of grey mullet, suggests that exogenous factors may also influences the grey mullet gut microbiota. To test whether migration history is associated with differences in the structure of grey mullet gut microbial composition, we compared our fish gut microbiota with historical seawater microflora of 33 seawater samples from previous studies (Supplementary Table 3)18,19,20,21. The historical seawater data were collected in the East and South China Seas along the migratory tracts of three cryptic species in the grey mullet species complex suggested by Shen et al.15.
Our analysis at the phylum level found that the gut microbial composition of NWP1 and seawater-microbiota in the ECS had similar compositions; in particular, most were dominated by Proteobacteria, but not Actinobacteria or Firmicutes (Fig. 6). A dendrogram was used to show the Pearson correlation coefficient results, which were calculated to elucidate the correlation between the fish microbiota and seawater microbes. Clear clusters were identified between mullet gut microbial communities of AD_NWP1 samples and many sea water microbial compositions in ECS. In contrast, the gut microbiota of fish belonging to cryptic species NWP2 were correlated vigorously with seawater samples SCS_P1, SCS_P2, SCS_A3, SCS_A4, SCS_S12 and ECS_T24, the bacterial communities of which were all dominated by Actinobacteria (Fig. 6, Supplementary Fig. 5). The dendrogram did not show any close clustering between gut microbial compositions in adult NWP3 and historical seawater data.
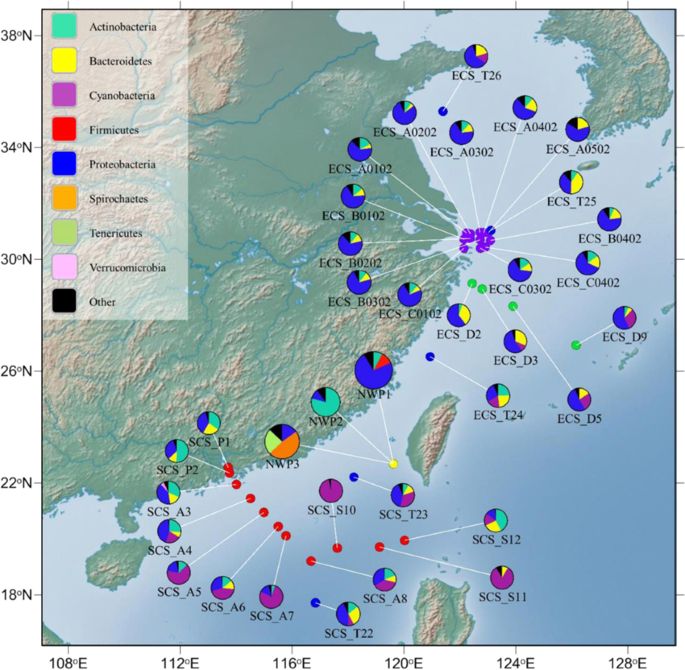
Map of collected fish gut microbiota and historical seawater microflora. The pie charts show the relative abundance of microbiota in collected adult grey mullet gut samples (yellow point) and seawater samples including 33 seawater samples published from four previous studies: Dong et al., 2013 (green points); Zhang et al., 2014 (red points); Zheng et al., 2016 (blue points), and Wu et al., 2017 (purple points). The colours in the pie chart present the relative abundance of different bacterial phyla, listed in top-left box, in each individual seawater sample or the grey mullet cryptic species. Name of fish groups and seawater samples are under their respective pie chart. NWP1, NWP2, and NWP3 indicate adults of three cryptic species of grey mullet M. cephalus in the Taiwan Strait. The map was created in GenGIS software58. Detailed information on seawater samples are provided in Supplementary Table 5.
Source: Ecology - nature.com
