Large-scale meta-analysis on food and human microbiomes
We performed a large-scale meta-analysis on microbiomes from food sources and human body sites to investigate the prevalence and diversity of LAB species in the human microbiome and their overlap with species and strains found in food. To achieve this goal, we considered 303 food metagenomes (152 publicly available and 151 obtained in this study) (11 datasets; Table 1 and Supplementary Data 1) that we curated in this study, which corresponded to different types of fermented foods and beverages8,9,10,11,12,13,14. In addition, we considered 9445 human metagenomes from 47 public datasets spanning multiple body sites (84% from the gut), age categories, countries, and lifestyles, which we retrieved from recent meta-analyses15,16.
Variable prevalence of LAB in the human gut
We considered reference-based taxonomic profiles17 of all 9445 human metagenomes15,16 (see “Methods”) and focused specifically on LAB species in this study (Supplementary Data 2). We detected 152 species belonging to the Lactobacillales order occurring in at least one of the metagenomes with a relative abundance >0.01%. Among them, we identified 70 species belonging to the LAB group and restricted the following analysis to the 30 of them having a prevalence >0.1% in the human gut (see “Methods”). These represented mainly species (spanning Lactobacillus, Lactococcus, Leuconostoc, Streptococcus, and Weissella genera) of potential food origin, including bacteria occurring in probiotic supplements, in addition to typically non-food origin species such as Lactobacillus mucosae, Lactobacillus ruminis, and Lactobacillus salivarius (Fig. 1). The two most prevalent species in the gut were Streptococcus thermophilus (prevalence 31.2%, i.e., present at >0.01% relative abundance in 31.2% of the gut metagenomes) and Lactococcus lactis (16.3%), both commonly found in dairy products (Fig. 1, Supplementary Fig. 1, and Supplementary Data 3). Multiple Lactobacillus species of predominantly food origin were detected at lower prevalence (3–5%) and comprised Lactobacillus casei/paracasei, Lactobacillus delbrueckii, Lactobacillus fermentum, and Lactobacillus rhamnosus). Non-food origin bacteria were also identified at remarkable levels such as Lb. ruminis (11.0%), Lb. salivarius (4.7%), and Lb. mucosae (4.0%). Although prevalence was variable, average relative abundance (computed on positive samples only) of single species was generally rather low (<2%), including the case of the two most prevalent species S. thermophilus (0.6%) and Lc. lactis (0.4%). Exceptions (rel. ab. >2%) were verified for Lactobacillus amylovorus, Lactobacillus brevis, and Lactobacillus buchneri, which however rarely occurred (prev. <1%).

We report the 30 LAB species having a prevalence >0.1% in the human gut. Values are obtained from 9445 publicly available human metagenomes and stratified by multiple host conditions (i.e., body site, age category, westernized lifestyle, and continent). Age category, westernized lifestyle, and continent statistics refer to stool samples only. Food results are obtained from 303 food metagenomes. Numbers and p-values (Fisher’s test, false discovery rate correction) in Supplementary Figs. 1–4 and Supplementary Data 4. Relative abundances in Supplementary Data 2 and 3.
Strong age-related patterns were verified for some of the species prevalent in gut samples (N = 7907) (Fig. 1, Supplementary Fig. 2, and Supplementary Data 4). S. thermophilus increased in prevalence from newborns (8.4%) to adults (33.7%, p < 1e − 40), with comparable average abundance. This may reflect the increase in consumption of yoghurts and other dairy products that can be sources of S. thermophilus18. A similar pattern was observed for Lb. delbrueckii (p < 1e − 10) and the non-food origin species Lb. mucosae (p < 1e − 10), Lb. ruminis (p < 1e − 20), and Lb. salivarius (p < 1e − 10), which suggests their gut colonization later in age. Also, Lc. lactis had higher prevalence in adults (15.8%) than newborns (8.6%, p < 1e − 6), with its detection in only one infant cohort originating from Estonia, Finland, and Russia19. Other lactobacilli were more prevalent and abundant in newborns such as Lb. casei/paracasei (p < 1e − 20 with respect to adults), Lactobacillus gasseri (p < 1e − 7), Lactobacillus plantarum (p < 1e − 4), and Lb. rhamnosus (p < 1e − 70). These species have also been detected in human breast milk20, suggesting their possible transmission from mother to infant through breastfeeding, as previously reported for Lb. plantarum21. Notably, these species were not found to be vertically transmitted from other mother’s body sites22.
Overall, we found that LAB are a subdominant component of the gut microbiome, although several species exhibited non-negligible contributions. More specifically, we identified 21 LAB occurring with prevalence >1% and 18 with relative abundance >0.5% when detected in the gut. It is reasonable to hypothesize that those species may be short- or long-term colonizers of the human microbiome.
Occurrence and abundance of LAB is linked to lifestyle
We then stratified the gut metagenomes in terms of host lifestyles (Fig. 1, Supplementary Fig. 3, and Supplementary Data 4), which revealed variations in prevalence and abundance between westernized and non-westernized populations for multiple species. Higher prevalence in westernized populations was observed for six lactobacilli, mostly of food origin, such as Lactobacillus acidophilus (p < 1e − 6), Lb. casei/paracasei (p < 1e − 4), Lb. delbrueckii (p < 0.01), Lb. gasseri (p < 1e − 6), Lb. rhamnosus (p < 1e − 9), and Lactobacillus sakei (p < 1e − 3). By contrast, Lb. mucosae (p < 1e − 8) and Lb. ruminis (p < 1e − 100) that do not occur in food were more prevalent in the non-westernized cohorts. Despite different patterns in terms of prevalence, all lactobacilli were on average more abundant in the westernized populations. Among the other genera, S. thermophilus was highly prevalent in the westernized cohorts (p < 1e − 50). Higher prevalence in the non-westernized group was observed for Lactococcus garvieae (p < 1 − e30) in addition to multiple heterofermentative species such as Leuconostoc citreum (p < 1e − 70), Leuconostoc lactis (p < 1e − 60), Weissella cibaria (p < 1e − 10), and Weissella confusa (p < 1e − 100), which is consistent with their widespread prevalence in raw vegetables23 that are likely consumed in such populations. In fact, non-western populations usually have hunter–gatherer diet and lifestyle, which is recognized to be characterized by high consumption of tubers, drupes, roots, and fruits24,25. Indeed, it was also reported that the!Kung and the Hadza, two non-Western African populations, still obtain 60–80% and 50–65% of their diet from plant foods, respectively26.
We further grouped metagenomes by host country of origin (see “Methods”) and identified more subtle geographical variations (Fig. 1 and Supplementary Fig. 4). Overall, food-associated lactobacilli were most prevalent and abundant in Europe, were less so in Asia and North America, and were almost absent in China (kept distinct from the other Asian countries due to its large sample size) and in the non-westernized populations. The higher prevalence in European cohorts was significant (p < 0.05) for Lb. casei/paracasei (8.0%), Lb. delbrueckii (6.6%, with a similar value in Asia), and Lb. rhamnosus (7.1%). Exceptions were Lb. gasseri, having comparable prevalence in continents including westernized cohorts, and Lb. fermentum, more prevalent in North America, South America, and China, with the latter observation being consistent with its widespread occurrence in Chinese fermented foods27. Non-food lactobacilli were not prevalent in Europe. Lb. mucosae exhibited high prevalence (>10%) in Africa, China, and South America, with comparable abundance across the globe. A similar trend was verified for Lb. ruminis, although with higher prevalence in non-westernized cohorts, whereas the presence of Lb. salivarius was distinctive for the Chinese population (p < 0.01). Among the other genera, Lc. lactis exhibited high prevalence across the entire globe (ranging from 11.5% in Africa to 44.4% in South America) with the sole exception of China (1.7%). S. thermophilus reached high prevalence in Asia (41.5%), Europe (39.6%), and North America (28.1%), but was much less prevalent in the Chinese (5.6%) and non-westernized (<3%) cohorts.
LAB species from food only partially match those in the gut
We established genome level links between the microorganisms populating the human microbiome and those found in food by integrating the genomes reconstructed from a set of 9445 human metagenomes with those from the set of 303 food metagenomes that we generated, collected, and curated in this work (Table 1 and Supplementary Data 1). More specifically, we considered 303 metagenomic samples spanning 11 datasets and coming from different types of cheese (N = 191), multiple fermented foods (N = 58), nunu (N = 20), milk kefir (N = 18), and yoghurt and dietary supplements (N = 16). We applied a validated16,28 computational pipeline that combined single-metagenome assembly, contig binning, and genome quality control to reconstruct de novo metagenome-assembled genomes (MAGs) from the set of food metagenomes (see “Methods”). We generated a total of 666 food MAGs (completeness > 50% and contamination < 5%) of sufficient quality according to previous recommendations29. These MAGs from food were integrated with the set of 154,723 MAGs that we retrieved from the 9445 human metagenomes using the same assembly-based pipeline16 and with the set of 193,078 reference genomes (available in GenBank as of March 2019). This resulted in a total of 348,467 genomes that were clustered at 5% genetic distance based on whole-genome nucleotide similarity estimation and recapitulated in species-level genome bins (SGBs, i.e., clusters of genomes spanning 5% genetic diversity, see “Methods”). The 666 food MAGs were grouped into 171 SGBs (Supplementary Data 5 and 6), which we discuss below on the basis of their occurrence in food samples and human gut (Fig. 2a, b).
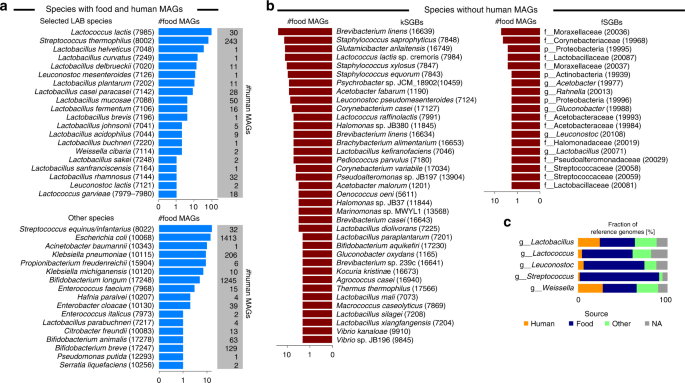
a Most prevalent species-level genome bins (SGBs) in 666 MAGs reconstructed from 303 food metagenomes and overlapping with human MAGs (i.e., found in at least one of the 154,723 human MAGs). Numbers in parenthesis represent the SGB IDs. b Most prevalent food SGBs not overlapping with human MAGs. kSGBs denote SGBs with at least one reference microbial genome, whereas fSGBs identify newly assembled SGBs from food metagenomes only. X-axes for a and b are in logarithmic scale. c Fraction of reference genomes per source type for the 30 selected LAB species and grouped by genera (the same plot at species-level is reported in Supplementary Fig. 6). Raw data in Supplementary Data 6 and 7.
Most of the food MAGs (349, 52.4%) belonged to SGBs also found in the human gut, with 265 of them associated with twenty of the thirty LAB species discussed previously (Fig. 2a top panel and Supplementary Fig. 5). The species most reconstructed from food sources was Lc. lactis (N = 90 MAGs), with 86 MAGs extracted from cheese. Sixty MAGs were associated with S. thermophilus, the majority of them was reconstructed from cheese and yoghurt, and five additional genomes were extracted from different fermented foods such as wagashi, beetroot kvass, ryazhenka, ruž’a, and labne. A consistent number of MAGs was also retrieved from Lactobacillus helveticus (33 MAGs from cheese), Lactobacillus curvatus (14 MAGs from cheese and 1 from sauerkraut), Lb. delbrueckii (11 MAGs from cheese or yoghurt in addition to single genomes from dietary supplement and tofu), Leuconostoc mesenteroides (5 MAGs from nunu and single genomes from bread kvass, ginger beer, milk kefir, beetroot kvass, ruž’a, and cheese), and Lb. casei/paracasei (4 MAGs from cheese, 2 MAGs from dietary supplements, and 2 MAGs from water kefir). We also extracted four MAGs of Lb. mucosae, a typical non-food microorganism that is usually found in the intestine of pigs or other animals30, and which we instead reconstructed from different fermented foods such as kimchi, kombucha vinegar, agousha, and sauerkraut.
We identified 17 additional non-LAB SGBs having MAGs from both food and human metagenomes, for a total of 84 food MAGs (12.6%; Fig. 2a bottom panel) and spanning three phyla (namely Actinobacteria, Firmicutes, and Proteobacteria). Some of these may be microbial contaminants in the food chain that can arise from different sources including animal, feed, and soil31,32. The SGB with the most MAGs (N = 16) was that containing Streptococcus equinus and Streptococcus infantarius genomes, two species usually found in the rumen33 but occasional pathogens for humans34, and which we found in African fermented foods13.
The majority of the food SGBs (134 out of 171), accounting for 317 MAGs (47.6%), did not exhibit an overlap with human MAGs, likely representing species unable to reach the colon or characterized by low prevalence and abundance in the human gut (Fig. 2b). Among them, 71 SGBs (53.0%; comprising 225 MAGs) contained at least one reference genome (kSGBs; Fig. 2b left panel). The most prevalent food-specific species was Brevibacterium linens (24 MAGs), which was reconstructed from multiple cheese types (i.e., surface ripened8, smear ripened14, hard, and tomme). Food-specific SGBs also included Staphylococcus saprophyticus (13 MAGs), Glutamicibacter arilaitensis (12 MAGs), and 58 MAGs from 21 LAB species spanning 6 families, the most prevalent being Lc. lactis subsp. cremoris. This set of MAGs and reference genomes showed a >5% genetic distance from Lc. lactis subsp. lactis genomes35, which we kept as a separate SGB (ID 7985) and found to be prevalent in both food and human metagenomes, in contrast to Lc. lactis subsp. cremoris, which was only detected in food metagenomes. Similarly, Lactococcus raffinolactis was divided into two SGBs, with human and food MAGs grouped in the SGBs 7989 and 7991, respectively.
Out of the 134 SGBs not overlapping with human MAGs, 63 SGBs (47%; comprising 92 MAGs) consisted of MAGs reconstructed in this study from food metagenomes without any reference genomes. These represented new species currently not represented in public repositories (Fig. 2b right panel), of which only 12 were assigned to known genera, and which should be targeted for cultivation-based analysis.
The set of genomes reconstructed and the SGBs identified in this study and that we made publicly available (see “Methods”) facilitated a more in-depth comparative genomics analysis.
Comparative genomics suggests a food origin for the gut strains
Within the available set of MAGs and reference genomes, we performed strain-level comparative genomic analysis for the set of 348,467 genomes previously described and comprising 193,078 reference genomes, 154,723 human MAGs, and 666 food MAGs. The 2859 genomes (including 1042 MAGs) associated with the thirty LAB species of interest were kept for comparative genomics purposes. To inform the comparative analysis, we retrieved and manually curated the source types for all genomes (see “Methods”) and grouped MAGs and reference genomes in three categories: human, food, and other. Genomes for which this information was missing were labelled as NA (7.9% of genomes; Fig. 2c, Supplementary Fig. 6, and Supplementary Data 7).
Overall, two-thirds of the reference genomes came from food (43.8%) and human sources (21.0%). The group of genomes from strains not isolated from foods or humans (22.8%) comprised 67 genomes from probiotics and dietary supplements in addition to 347 genomes mainly coming from animal sources. The proportions of species assigned to the different source types was quite variable across species, with a general under-representation of human genomes corresponding to LAB that were prevalent in non-westernized cohorts (Fig. 2c and Supplementary Fig. 6). This reflected the overall scarce availability of genome from isolates for a substantial fraction of the non-pathogenic, commensal members of the human microbiome as recently highlighted16,36,37. Reference genomes from human samples were surprisingly almost absent in the case of prevalent species such as Lc. lactis (with only one reference genome from the vagina and one MAG from the gut) and S. thermophilus (with only one MAG from the gut). The absence of good reference genomes in public repositories prevented the comparison of food and human strains until now, which we aimed to overcome in the present study through an extensive comparative genomics analysis.
S. thermophilus was the species of LAB most frequently reconstructed from metagenomes (243 human and 60 food MAGs; Fig. 3a), an observation consistent with its high prevalence from mapping-based taxonomic profiling (Fig. 1). Comparative genomics, also including 44 reference genomes, did not highlight food-specific or gut-specific sub-clades, suggesting that food can be regarded as the main source of this species in the human microbiome. S. thermophilus also appeared to be a quite genetically diverse species both in food and human sources with MAGs reconstructed from Asian gut metagenomes enriched in a specific clade (Clade A, Fig. 3a, p < 1e − 10). Lb. delbrueckii was not prevalent in the gut, and the only two subspecies found in human samples were subsp. lactis and subsp. bulgaricus (Fig. 4a). Human MAGs of both subspecies clustered together with food MAGs and isolates, again indicating food as the most likely source of this species in the gut. On the other hand, subsp. delbrueckii, subsp. sunkii, and subsp. jakobsenii were found in food, but never reconstructed from the gut. Although Lb. rhamnosus was the LAB species for which the greatest number of genomes corresponding to human isolates (N = 105) was available, we collected only 32 human MAGs, which is in agreement with its low prevalence and abundance in the gut (Fig. 4b). We identified a specific cluster including 17% of the Lb. rhamnosus human genomes that included the reference genome associated with the Lb. rhamnosus strain GG (LGG), which may be due to recent consumption of commercial products due to its wide use in probiotic supplements38.
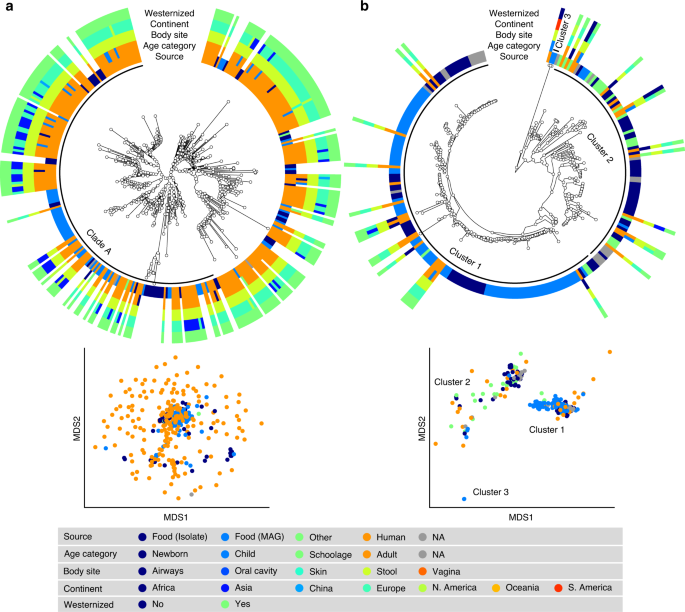
a S. thermophilus is a genetically diverse species both in food and human sources with MAGs reconstructed from Asian gut metagenomes enriched in Clade A (p < 1e − 10). b Lc. lactis subsp. lactis is formed by three main clusters: Cluster 1 exhibits an overall low diversity and includes mostly food genomes related to cheese and dairy fermentation; Cluster 2 is dominated by environmental and raw vegetable products and more diverse human MAGs; Cluster 3 includes only two MAGs from nunu. Phylogenetic trees were built on species-specific marker genes and report five different metadata. Multidimensional scaling (MDS) on average nucleotide identity (ANI) distance is coloured with source information.
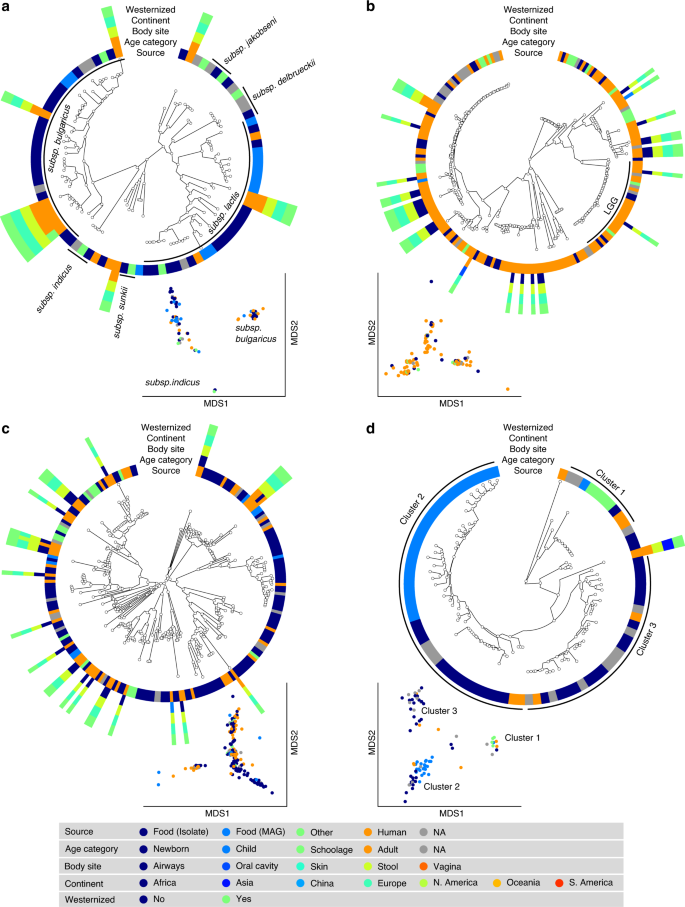
a Lb. delbrueckii is not prevalent in the gut, and the only two subspecies found in both food and human samples are subsps. lactis and. bulgaricus. Subsps. delbrueckii, sunkii, and jakobsenii are found in food, but never reconstructed from the gut. b Lb. rhamnosus exhibits the greatest number of genomes from human isolates but is scarcely reconstructed from metagenomes. A specific cluster identifies the LGG strain. c Lb. casei/paracasei includes reference genomes identified as both Lb. casei and Lb. paracasei. We detect two main clusters both occurring in food and human samples. d Lb. helveticus exhibits three main clusters, with Cluster 1 including all the dietary supplement strains (source in green), while food genomes are predominantly spread across the other two groups. Phylogenetic trees were built on species-specific marker genes and report five different metadata. Multidimensional scaling (MDS) on average nucleotide identity (ANI) distance is coloured with source information.
The highest number of food MAGs was obtained for Lc. lactis (N = 90, Fig. 3b). We refer here to subsp. lactis, whereas subsp. cremoris was associated with 12 food MAGs but never reconstructed from human metagenomes. Lc. lactis subsp. lactis formed two distinct clusters including both food and human genomes. The first cluster included 63% of the genomes, exhibited an overall low diversity (<0.8% genetic distance between closest genome pairs), and included all the food genomes related to cheese and dairy fermentation. The second cluster was more diverse, dominated by environmental and raw vegetable products, and included the only MAG from human skin and the three gut MAGs from non-westernized cohorts. An additional cluster containing two genomes from nunu13 was never found in humans and exhibited a >3% genetic diversity from all other genomes. Such results highlighted the overall importance of conducting strain-level analysis on the food-gut axis, depicted here by the identification of two main clusters in the human gut associated with different food sources (i.e., one from cheese and dairy fermentation, and the other one from environmental and raw vegetables products). Strains of these clusters are likely characterized by differences in functional traits and potential interaction with the host that deserve to be investigated in future studies.
The SGB 7142 (N = 216, Fig. 4c), labelled Lb. casei/paracasei, included reference genomes identified as both Lb. casei and Lb. paracasei, which, as recently highlighted, can be used interchangeably39. Within the combined species, we detected two main clusters, both of which occurred in food and human samples. The major cluster contained 86% of the available genomes, including all the dietary supplement strains and the majority (86%) of the human MAGs. Consistent with its low abundance (Fig. 1), only seven reference genomes and a single MAG were reconstructed from human samples for Lb. helveticus (Fig. 4d). We identified three main subspecies, all occurring in both food and human sources. One cluster included all the dietary supplement strains, whereas genomes coming from food were predominantly spread across the other two groups.
Despite the high number of collected genomes (N = 369), Lb. plantarum was scarcely prevalent (1.8%) and abundant (av. 1.2%) in the gut (Fig. 1), which was reflected by only 11 MAGs being reconstructed from human microbiomes (Supplementary Fig. 7). All of these belonged to the main cluster (96% of the total genomes) associated with subsp. plantarum. A separate cluster was identified as subsp. argentoratensis, which was found in both food and human isolates but never reconstructed from metagenomes. The occurrence of multiple subspecies within the same SGB was also observed for eight additional LAB, i.e., Lb. brevis, Lb. fermentum, Lactobacillus johnsonii, Lactobacillus reuteri, Lb. sakei, L. lactis, L. mesenteroides, and W. cibaria, (Supplementary Fig. 7). On the other hand, Lc. garvieae was spread into two different SGBs, with one comprising human MAGs from both westernized and non-westernized populations and the other only from non-westernized cohorts (Supplementary Fig. 7). No genomes from food samples were collected at all for Lactobacillus crispatus, Lb. gasseri, Lactobacillus jensenii, Lb. ruminis, and Lb. salivarius (excluding a single isolate from ground beef). The non-food species Lb. ruminis and Lb. salivarius were quite prevalent in the gut with 145 and 42 MAGs reconstructed from human metagenomes, respectively (Supplementary Fig. 7). For both species, isolate and MAGs extracted from the gut were distinct from genomes isolated from other animal microbiomes, which suggested long-term adaptation of these species to the human gut. We also identified a specific Lb. salivarius cluster associated with dietary supplement strains, which was found in a couple of saliva samples but never in the human gut.
LAB occurrence in non-human primates is affected by captivity
We finally considered the set of 203 publicly available gut metagenomes from non-human primates (NHPs) that was recently retrieved, curated, and processed with the same pipeline employed in this study28. It comprised 22 host species from 14 different countries in five continents. Among the 2985 reconstructed MAGs, we found that only 46 of them (1.6%) were assigned to the Lactobacillales order (Supplementary Data 8), which suggested an overall low prevalence and abundance of LAB in the NHP gut microbiome. We found strong differences between MAGs retrieved from wild NHPs and those extracted from NHPs living in captivity. Wild NHPs generated 29 MAGs of LAB, with 66% of them associated with new species not available in public repositories and never found in human metagenomes, therefore likely representing bacteria peculiar to the NHP gut microbiomes. Ten MAGs were instead associated with kSGBs, with only five of them belonging to LAB species found also in human gut metagenomes such as Lc. garvieae (N = 3), Lc. lactis, and W. cibaria. Comparative genomics analysis highlighted that the strains harboured in NHPs were quite different from those reconstructed from human microbiomes (Supplementary Fig. 8). Interestingly, the three MAGs of Lc. garvieae resembled more the strains found in non-westernized human populations in terms of nucleotide identity. No MAGs from lactobacilli were extracted at all from wild NHPs. A very different situation was observed in captive NHPs (Supplementary Fig. 8), in which the 17 MAGs were exclusively reconstructed from kSGBs associated with multiple Lactobacillus species, i.e., Lb. acidophilus, Lactobacillus animalis (N = 2), Lb. johnsonii (N = 4), Lb. mucosae (N = 2), Lb. reuteri (N = 5), and Lb. salivarius (N = 3). Strains of Lb. reuteri and Lb. salivarius found in NHPs were distinct from those extracted from human and food sources, which suggested possible host adaptation mechanisms. A stronger overlap among NHPs, human, and food MAGs was instead observed for the other species and likely linked to the sharing of strains due to the exposition of NHPs living in captivity to human-like environments and diets40.
Source: Ecology - nature.com
