Field sampling
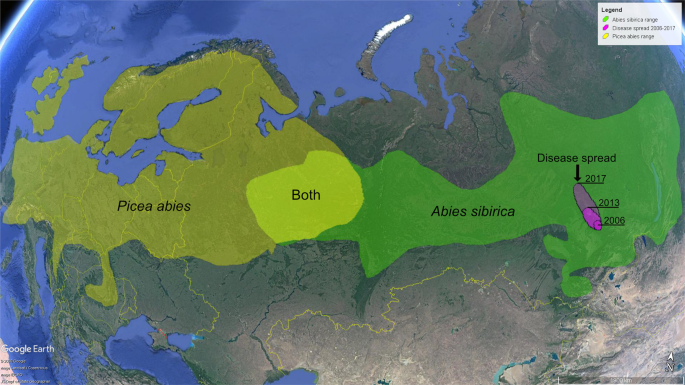
Investigated site was located in Central Siberia, about 15 km west of Krasnoyarsk (N56°01′41.46″; E92°34′39.30″). Samples were collected from five A. sibirica trees exhibiting typical symptoms of the disease. The trees were approximately 30–50 years-old, 6–13 m high, and 8 –14 cm in dbh. The sampling targeted parts of branches and stems representing discrete cankers (Fig. 2A–C). Symptomatic parts of the branches were cut off using a handsaw. Cut branch sections were approx. 4 cm long and 2 cm in diameter. Samples from stems were taken directly as pieces of diseased bark and wood using a chainsaw and an axe. Approximate size of stem samples taken in the field was 10 × 5 × 3 cm. Each sample was individually placed into a plastic bag and transported to the laboratory. A total of 64 samples was collected from three arbitrary categorized symptomatic canker types (Table 1).
Isolation of mycelia and morphological identification
In the laboratory, phellem was removed from the surfaces of cankers, and 20 × 5 mm size slivers of phloem/xylem were aseptically cut off across the canker-symptomatic areas, and sampled using ethanol + flame sterilized scalpel and forceps, taking 5–7 slivers per canker. Numbers of sampled cankers from each symptomatic category are shown in the Table 1. The slivers were surface sterilized in 70% ethanol, followed by rinsing in sterile water, all for 30 seconds per step, placed on 2% malt extract agar (MEA) and incubated at 20 °C in the dark. Mycelia that emerged from the samples were cut off from uniform morphologically discrete hyphal colonies and subcultured onto fresh MEA.
Morphological observations of the colonies in culture were carried out on isolates grown on potato-dextrose agar (PDA) (Fig. 2D), carrot agar (CA) (Fig. 2E), MEA (Fig. 2F), carnation-leaf agar (CLA), and slow nutrient agar (SNA) for 4 wk. at 20 °C with alternating 12 h/12 h mixture of cool-white fluorescent + UV-light/darkness. The colony growth rate was measured on 9 cm Petri dishes containing PDA, 5 mm deep, inoculated with a 10 mm diameter mycelial plug after 14 days. Microscopic observations of the isolates were made using an Olympus CX41 microscope (Olympus Co., Tokyo, Japan) and a scanning electron microscope Hitachi SU3500 (Hitachi, Tokyo, Japan). Fourteen pure cultures showed morphological characters typical for Acremonium-like (Corinectria) anamorphs (Table 1; Fig. 2D–F; Fig. 3)2,3, and four of those were included into further molecular analyses.
Molecular identification and phylogenetic analyses
DNA isolation from the four Siberian cultures was done using the CTAB method18. Briefly, fungal mycelium of each strain was collected from Petri dishes into individual 1.5 ml centrifugation tubes and together with glass beads homogenized by vortexing. To each tube, 1 ml CTAB buffer (3% cetyltrimetylammoniumbromide, 2 mM ethylenediamine tetraacetic acid, 150 mM Tris–HCl, 2.6 M NaCl, pH 8) was added, and the samples were incubated at 65 °C for 1 h. After centrifugation, the supernatant was sequentially extracted with an equal volume of chloroform, centrifuged for 7 min at 13,000 rpm, precipitated with 1.5 volumes of 2-propanol, washed with 70% ethanol, dried and redissolved in 50 μl ddwater18. Concentration of DNA in individual samples was determined using a Nanodrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
The fragments amplified were α-actin (ACT), β-tubulin (BTUB), the complete internal transcribed spacer of rDNA (ITS) and translation elongation factor 1α (TEF1). Primers and protocols for amplification were as in González and Chaverri2. The corresponding reference sequences for Corinectria species, Neonectria faginata, N. neomacrospora, and Thelonectria discophora were downloaded from the GenBank and are listed in Table 2. The sequences of each genetic marker were aligned using ClustalW and manually adjusted in BioEdit19. A partition homogeneity test in PAUP* 4.0a16620 has been run to check if there were no conflicts among different loci. As no significant conflict was detected (p > 0.45), DNA sequences were concatenated into a single supermatrix. The JModeltest21 was used to determine the nucleotide substitution model for the combined dataset, which was GTR + G. The Bayesian Inference (BI)-based phylogenetic approach was performed in MrBayes v3.2.722. Support for nodes was estimated using 1,000 fast bootstrap (BS) pseudo-replicates in PAUP Maximum likelihood (ML) analyses and by the posterior probabilities of nodes calculated after convergence of two independent MrBayes runs each of two chains run to 2 × 106 generations, sampling every 1,000 generations with a consensus tree computed after a final burnin of 20% of the trees. Bayesian posterior probabilities ≥0.9523, and ML bootstrap support ≥70% were considered to be significant. Sequences of Siberian isolates generated were deposited in GenBank (Table 2).
Pathogenicity tests
Two isolates of Acremonium-like (Corinectria) anamorph N1 (NfP5.7) and N2 (NfSp3.4) were tested for pathogenicity. Prior to the experiment, the isolates were stored in the lab on PDA for one year under 4-6 °С. Pathogenicity tests involved a total of 90 saplings (30 and 30 for each respective isolate, and 30 for control) and 900 seeds/subsequently seedlings of A. sibirica, and 900 seeds of P. abies (here, in a similar setting for both tree species, 300 + 300 for each isolate, and 300 for control) (Table 3). Control plants/seeds were subjected to similar test procedures, but in the absence of the anticipated Acremonium-like (Corinectria) pathogen. Seed material was collected from randomly selected trees of the respective species. Comparisons of recorded data between inoculated/control sets were made using chi-squared tests (for proportions) and t-tests (for means).
Saplings of Abies sibirica
In this part of the work, methodological approach followed studies by Bakys et al.24,25, where artificial inoculations were used to test pathogenicity of fungal isolates to saplings of Fraxinus excelsior L. Saplings of A. sibirica (age 7–10 years, height 40–45 cm, diameter at root collar 10–12 mm) were inoculated with wood chips, size 20 × 5 × 3 mm (length × width × thickness) pre-colonized by mycelium of a tested isolate. For this, sterilised chips were placed on a pure culture for approx. 4 weeks. Bark wounds were cut on stems approx. 50 mm high from the soil, the inoculated chip placed on a wound and wrapped with Parafilm (Bemis Company Inc, Neenah, WI, USA). Control saplings were of the same characteristics and were treated in a similar manner, but using sterile chips. Each sapling was planted in a container of 20 litres sterilised sand, placed in a climatic chamber at 25 °C and subjected to periodic humidification under natural light for three months (March, April, May). The following data were recorded.
- 1.
Incidence of dead and diseased saplings per isolate-inoculation set, and control set.
- 2.
The viability of bark and phloem, scored as either visually healthy (bark tightly attached, phloem colour light-green), or diseased/necrotic (bark detached, phloem colour brown to black).
- 3.
Extent of necrosis in cambial zone beyond inoculation wound (Fig. 5). It was measured with a flexible ruler: longitudinal extension from the inoculation boundary both upwards and downwards, and lateral at the cross-section to both sides, left and right, making a total of four measurements per inoculation wound.
- 4.
Re-isolation frequency of inoculated fungus from dead and diseased saplings. At first, presence/absence of the mycelium was visually evaluated at 5-10 mm distance above from the inoculation point by removing fragment of bark with a sterile scalpel and placing it into a “wet camera” (Petri dish containing double layer of sterile filter paper soaked in distilled water), and subsequently checking for fungal growth with the use of a microscope. When present, a fragment of mycelium was removed using sterilised needle and placed on CA and MEA to record an appearance/morphology of mycelial growth characteristic for inoculated Acremonium-like (Corinectria) isolate (Fig. 2E, F; Fig. 3). All inoculated saplings (30 + 30) were subjected to re-isolations (Table 3).
Seeds and seedlings of Abies sibirica
This part of the work followed modified methodological approach used in studies by Lazreg et al.26, Alghuthaymi et al.27, and Güney & Güldür28. Seeds of A. sibirica were sterilised in 0.5% dilution of KMnO4 for 120 min, washed with sterile water, dried in-between layers of sterile filter paper and sawn into plastic 350 ml volume containers, size 107 × 82 × 70 mm (length × breadth × height), filled with heat-sterilised sand intermixed with wood chips of size 20 × 10 × 3 mm. The chips were sterilised and pre-colonised by the mycelium by placing them on a pure culture of the inoculum fungus for approx. 4 weeks. At first, a bottom of each container was covered by a 5 mm thick layer of wet sterile sand, on the surface of which were placed pre-colonised chips, fifteen in each container. Placement scheme was in a straight-angled rows 5 × 3: five chips in three rows; distance between chips in a row, approx. 1.5 mm, distance between rows approx. 12 mm. They were covered by 2 cm thick layer of wet heat-sterilised sand. Seeds were sown on sand surface and afterwards thinly shed by sand, 100 seeds per container, making in total three containers per each of the two tested strains of the fungus (300 seeds/strain, in total 600). Three control containers were processed in a similar manner, but included sterilised chips (Table 3). All containers were covered by caps (for approx. 25 days, after which regular germination has appeared and caps were removed), placed in a climatic chamber at 25 °C and subjected to periodic humidification under natural light for three months (under similar regime as for saplings, see above). The following data were recorded.
- 1.
Frequency of seed germination.
- 2.
Incidence of dead and diseased seedlings.
- 3.
Extent of chip surface coverage by inoculated mycelium (exhibiting typical yellow – orange color) by the end of the experiment. It was evaluated visually, according to the following scoring system. Score 4: mycelium covers the surface of an inoculated chip by >75%; Score 3: 50–75%; Score 2: 25–50%; Score 1: <25%; Score 0: mycelium absent (as, e.g., in controls that also have been checked). Each inoculated chip has been subjected to such evaluation, meaning 15 from each container, thus 45 for each isolate (Table 3).
- 4.
Re-isolation frequency from inoculated chips. This was done by cutting off (sterilised scalpel) approx. 5 × 5 mm size fragment of a chip, and then transferring (sterilised forceps) cut wood sample to a “wet camera”. After 3–6 days, emerging mycelia from those were individually transferred (sterilised needle) to Petri dishes containing MEA. From each inoculated container, eight chips were subjected for re-isolation, making 24 for each isolate, a total of 48 (Table 3).
- 5.
Re-isolation of the pathogen from dead and diseased seedlings. The seedlings were extracted from the container soil with roots, individually washed under the running water, then submerged for 1 min in 5% dilution of Ca-hypochlorite, washed in sterile water, dried with sterilised filter paper, cut (using sterilised scalpel) into 15–20 mm fragments, which were placed into Petri dishes onto MEA and CA. All dead and diseased seedlings from each container were subjected to re-isolation, 169 for the isolate N1 and 165 for N2, making a total of 334 (Table 3).
Seeds and seedlings of Picea abies
This part of the work followed modified methodological approach used in the study by Alghuthaymi et al.27. Each of the same tested two Acremonium-like isolates were grown separately in two separate sets in liquid Norkrans media during 14 days at 25 °С. In order to test for germination rates and subsequent condition of eventually germinating seeds, 300 + 300 seeds of P. abies were soaked in the cultural filtrate of each respective strain for 24 hours, and placed to germinate in sterile Petri plates with double layer of moist filter paper (“wet cameras”) for 21 days at a temperature of 25 °C. For control, 300 seeds were soaked in sterile water and subjected to the germination test in a similar way (Fig. 6). The following parameters were recorded.
- 1.
Frequency of seed germination.
- 2.
Incidence of dead and diseased seedlings.
- 3.
Length of a seedling stem and main root.
- 4.
Seedling weight. This was done by individually removing of survived seedlings from”wet camera”, dried till air-dry permanent weight, and then weighted.
- 5.
Re-isolation of inoculated fungi from seedlings was performed by individually washing those under the running water, then submerging for 1 min in 5% dilution of Ca-hypochlorite, washed in sterile water, dried with sterilised filter paper, and placing those into Petri dishes onto MEA and CA.
Source: Ecology - nature.com
