The microcosm experiments were conducted with 2000 m deep water samples from the western Pacific Ocean, a typical oligotrophic gyre (Fig. 1 and Supplementary Fig. 1). The in situ prokaryotic and viral abundances decreased from 0.70 and 6.17 × 106 particles mL−1 in surface waters to 0.49 and 1.70 × 106 particles mL−1 at 2000 m. With a series of filtrations and dilutions of water from 2000 m, microcosms were set up with the prokaryotic community either exposed to viruses (+virus) or with a reduced viral abundance (−virus) (Fig. 1). In order to investigate the development of deep sea bacterioplankton and avoid the possible limitation for bacterial growth which usually occurred in in situ deep sea environments (e.g., see refs. 1,14), prokaryotes were diluted at the beginning of the incubation. The initial prokaryotic abundances were 1.5 × 104 particles mL−1 and 2.1 × 104 particles mL−1 for −virus and +virus treatments, respectively (Fig. 2a, Supplementary Data 1). Although the filtration and dilution steps may produce slight differences between treatments (e.g., the amount of organic matter), reducing the bacterioplankton population size in our experimental setup should have alleviated this influence, and organic matter was likely not a limiting factor in our incubation system15,16,17. Resource competition among the bacteria would also have been relieved by dilution, as fewer bacteria would have been competing for the same amount of nutrients. For the −virus treatments, viral abundance was reduced to 3.55 × 105 particles mL−1 at the beginning of incubation and showed small variations during incubation (Fig. 2b). After 12 days of incubation, viral abundance in the +virus treatments were higher and showed clear fluctuations, increasing from 5.83 × 105 particles mL−1 to 1.44 × 106 particles mL−1 (Fig. 2b). The virus-to-bacteria ratios in both treatments displayed similar temporal patterns and decreased after 5–7 days of incubation (Supplementary Fig. 2), which was caused by bacterioplankton growth in the −virus treatments and viral decay in the +virus treatments, respectively. Higher virus-to-bacteria ratios were always observed in the +virus treatments throughout the incubation, demonstrating the successful experimental setup with relatively strong top-down control by viruses of deep-sea bacterioplankton. In addition, compared with similar experiments for surface bacterioplankton and virioplankton10, a longer incubation time (12 days) was required for the deep-sea bacterioplankton and virioplankton in our study. This is consistent with the relatively low bacterial and viral activity, and then longer bacterial and viral turnover time, recorded in deep sea1. For example, the viral turnover times in the deep North Atlantic Ocean were 11–39 days and the turnover times of the prokaryotic community were about 34–54 days2,3,4,6.
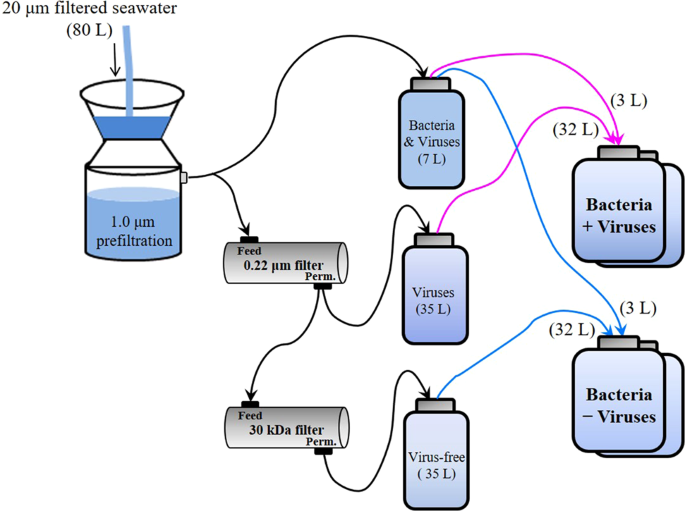
Schematic diagram of the experimental setup for deep sea bacterioplankton with normal and reduced pressure of viral lysis.
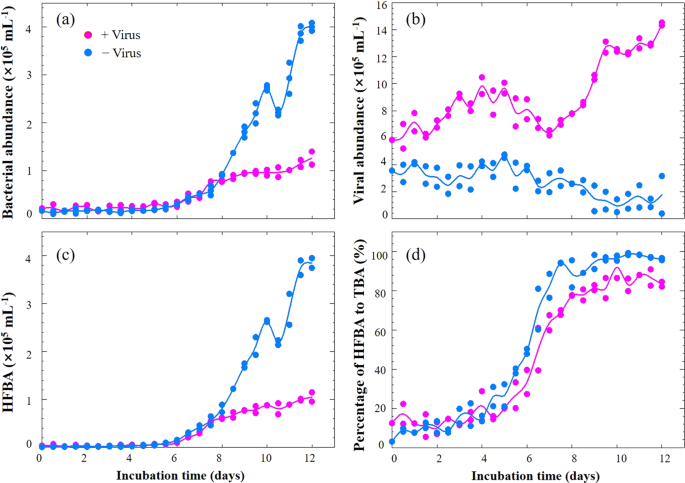
a Bacterial abundance, b Viral abundance, c High-fluorescence bacterial abundance, d Percentages of high-fluorescence bacterial abundance to total bacterial abundance. HFBA high-fluorescence bacterial abundance, TBA total bacterial abundance. Dots show two replicates at each time point and lines represent the average values.
Effects of viruses on deep-sea bacterioplankton abundance
The bacterial abundances in the +virus and −virus treatments showed a rapid growth between Days 5 and 7, raising to 0.48 × 105 particles mL−1 and 0.51 × 105 particles mL−1, respectively (Fig. 2a). Afterwards, the −virus treatments were characterized by more rapid growth of bacterioplankton, reaching a higher abundance, than the +virus treatments (Fig. 2a). After 10–12 days of incubation, the bacterioplankton abundance in the –virus treatments was 3.2 times higher than that in the +virus treatments (Fig. 2a). Concurrently, the abundance of viruses in the two treatments showed distinct trends: viral abundance increased in the +virus incubations but decreased slightly in the −virus incubations (Fig. 2b). The repressing effects on prokaryotic abundance in the +virus treatments demonstrated the presence of top-down control by viruses of deep-sea bacterioplankton. The reduction of viral abundance in the −virus treatment suggests decay of viral particles due to reduced encounter rates with hosts, hence resulting in lower viral infection rates.
Since it has been proposed that viruses mainly infect active hosts, e.g., cells with high RNA content18, we investigated the dynamics of high-fluorescence signal bacterioplankton, containing relatively high nucleic acids19,20, using flow cytometry (Fig. 3). For both treatments, the abundances of high-fluorescence bacterioplankton remained relatively stable until Day 6. But in the latter period (i.e., after Day 6), high-fluorescence bacterial abundance (HFBA) increased more rapidly in the −virus treatments than in the +virus treatments were observed in the latter period (Fig. 2c). For both treatments, the percentages of high-fluorescence bacterial to total bacterial abundance (TBA) ranged from 3.0 to 49.0% for the first half of the incubation and then exceeded more than 70% during the second half. The increasing trends of HFBA were similar to those of TBA, suggesting that the growth and development of potentially active bacterial assemblages in our incubation were mainly due to the cells containing high nucleic acids content. Lower percentages of high-fluorescence cells were observed in the +virus than in the −virus treatments, especially in the latter period (Fig. 2d). The repression of active cells caused by the presence of viruses accounted for most (70%) of the difference between the abundance of total bacterioplankton in the two treatments. Our finding suggests that viruses infect and lyse active bacterioplankton cells in the deep sea, which is consistent with observations of surface water18. In contrast, potentially less active bacterioplankton as indicated by low nucleic acid content might be less susceptible to viral infection or the infection processes are much slower21. Therefore, in the deep-sea environments, the majority of bacterioplankton might be slow growing defense strategists, while the potential winners of competition for nutrients would be controlled by viruses.
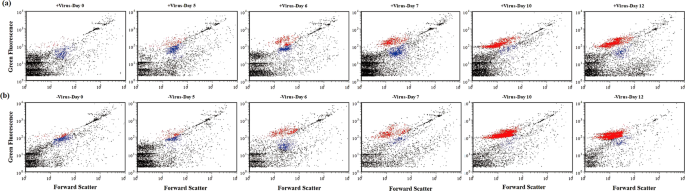
a Flow cytograms showing development of bacterioplankton with normal viral lysis pressure. b Flow cytograms showing development of bacterioplankton with reduced viral lysis pressure. Red: high-DNA bacteria; Blue: low-DNA bacteria.
Effects of viruses on deep-sea bacterial diversity and community structure
Bacterial diversity and community composition were investigated with 454 sequencing of PCR-amplified 16S rRNA genes at the DNA and RNA levels, respectively. After quality control and normalization, 64,427 sequences were obtained, and 975 OTUs were assigned based on 97% similarity of sequences. At the beginning of the incubation, the bacterial diversity (Shannon index) at the DNA level was 5.7. After 12 days of incubation, bacterial diversity decreased to 4.7 in the +virus treatments and to 2.0 in the −virus treatments (Fig. 4a). Similarly, the bacterial diversity revealed by RNA analysis decreased after incubation and was higher in the +virus treatments (Shannon: 3.9, Phylogenetic Diversity: 13.1) than in the −virus treatments (Shannon: 3.1, Phylogenetic Diversity: 10.3). Although rRNA is not simply related to bacterial activity, it has been used as tentative indicator for the active bacterial community or for an anticipatory life strategy, i.e., being able to respond quickly to environmental changes (e.g., see ref. 22). For the deep-sea, such environmental changes could include the mixing of water masses23, sinking particles, or the in situ formation of particles (e.g., see ref. 14).
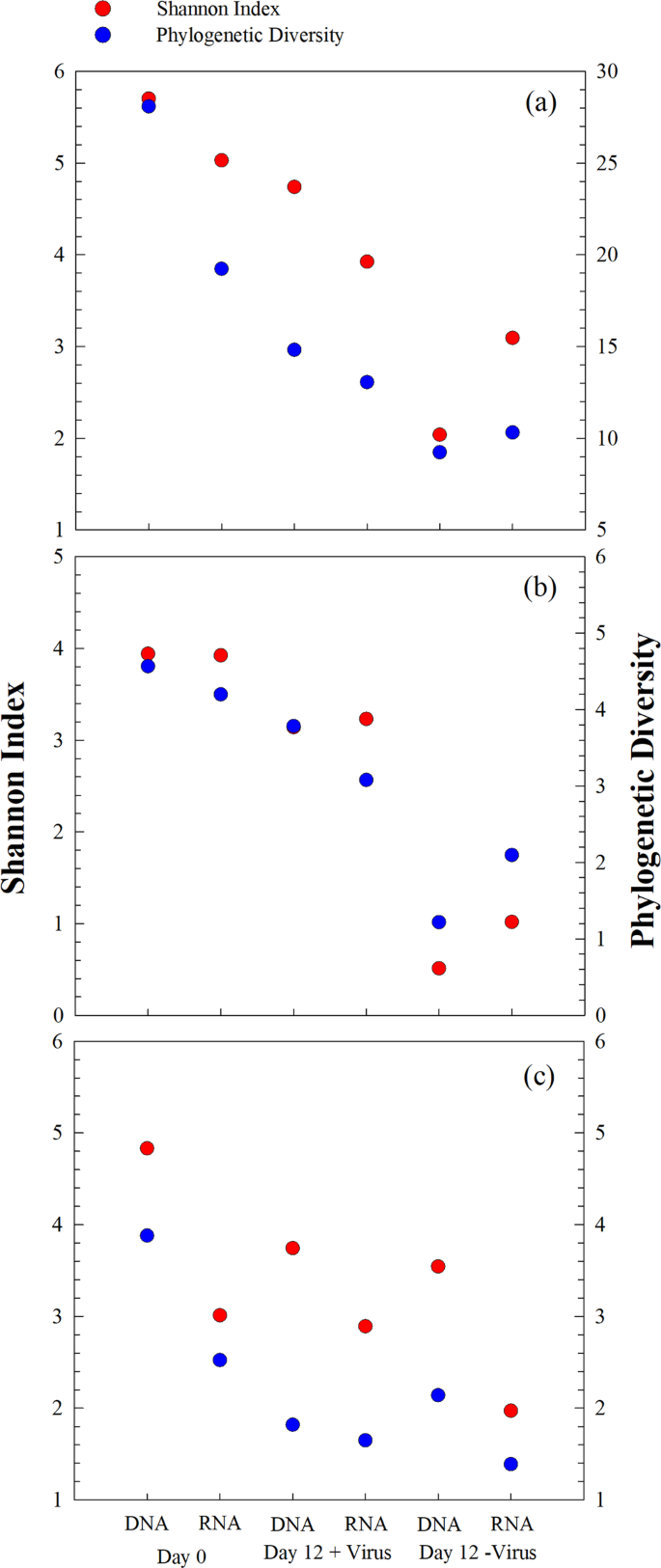
a Total and active bacterial diversity, b Total and active Alphaproteobacterial diversity, c Total and active Gammaproteobacterial diversity. Diversity is shown as Shannon index and phylogenetic diversity. Data show the mean of two independent incubations.
For the major phyla present at the beginning of the experiment (e.g., Alphaproteobacteria and Gammaproteobacteria), diversity generally decreased after incubation; moreover, higher diversity was always observed for the +virus treatment than for the –virus treatment at both the DNA and RNA levels (Fig. 4b, c). Previous studies have suggested that the presence of viruses supports higher bacterial diversity in surface oceans8,10. The mechanism behind this phenomenon could be explained by the “kill the winner” hypothesis, which states that viruses infect and lyse the bacterial winner of resource competition, prevent their blooming, and create niches for other bacteria24,25,26. This would allow for more bacterial species to be exit in a specific environment, leading to higher diversity. Furthermore, the release of organic matter during cell lysis alters the composition and bioavailability of organic nutrients, thus changing the composition of the bacterial community in surface waters27. Our study suggests that viruses play a similar ecological role in structuring the microbial community in deep-sea ecosystems.
At the beginning of the incubation, the RNA-based bacterial diversity was lower than the DNA-based, indicating that some bacterial lineages were inactive (or not anticipatory) in the original bacterioplankton community. In the Day 0 samples, Alphaproteobacteria showed relatively higher diversity at the RNA level than at the DNA level (Fig. 4b), suggesting that they are highly active in situ. The loss of Gammaproteobacteria diversity was greater than that of Alphaproteobacteria after incubation with viruses (Fig. 4b, c), contributing to the majority of diversity loss of total bacterioplankton, especially at the DNA level. Similar to the total bacterial population, higher diversity loss was observed for both Alphaproteobacteria and Gammaproteobacteria in the −virus treatments than in the +virus treatments. In addition, higher diversity of active bacteria compared with that of total bacteria was observed in the −virus treatments, which was due to the same trend occurring in Alphaproteobacteria (Fig. 4a, b).
To represent the major bacterial lineages across all samples, the 58 most abundant OTUs (accounting for 90% of all sequences after normalization) were selected. These major OTUs were affiliated with Proteobacteria, Bacteroidetes, and Cyanobacteria (Fig. 5). Cluster analysis showed that the +virus treatments displayed more similarity to bacterial communities in the −virus treatments than those in situ (Day 0 samples) (Fig. 5). However, at both the DNA and RNA levels, the similarities between the +virus treatments and in situ environment were higher than those between the −virus treatments and in situ environment (Fig. 5). This indicates that in addition to general influences on diversity, viruses also sustain the community structure of deep-sea bacterioplankton. Similar trends have been recorded in studies on surface water (e.g., see refs. 10,28).
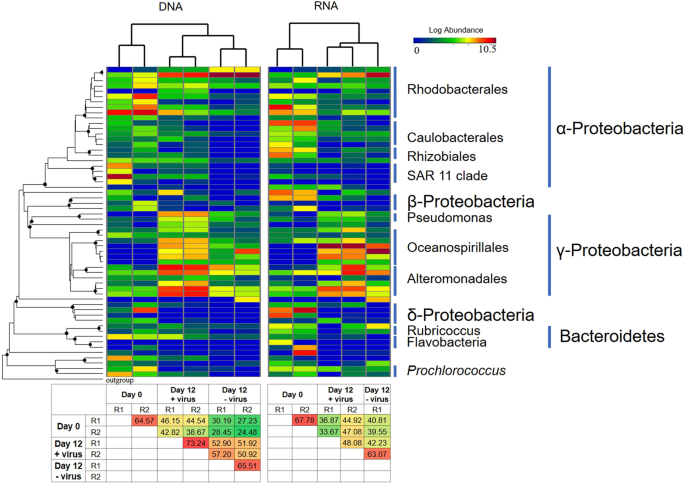
The cluster analysis of bacterial community structure was generated based on Bray-Curtis distances between samples. Heatmap shows the relative abundance (log [relative abundance + 1]) for each OTU. Phylogenetic tree (on left) was reconstructed by representative sequence from each OTU with Maximum-Likelihood method using FastTree. Filled circle on the nodes show bootstrap values higher than 50% (1000 resamplings). One archaeal sequence from Picrophilus torridus (AE017261) was used as outgroup. The two heatmaps on the bottom show the similarity (%) of Bray–Curtis distances between samples. Day 0 samples were collected before aliquoting into microcosms and considered as control for both +virus and −virus treatments.
Effects of viruses on the relative contribution of deep-sea bacterial lineages
The relative abundances of specific bacterial lineages (DNA level) and potentially active bacterial lineages (RNA level) were assessed. Before incubation, Alphaproteobacteria dominated the bacterial population at both the DNA level and RNA level (Fig. 6). After 12 days of incubation with viruses, Gammaproteobacteria dominated the potentially active bacterial community (rRNA level). Detailed examination of the bacterial taxa in our experiments showed that nine of the top ten abundant OTUs showed a difference between the +virus and −virus DNA libraries, including Sulfitobacter, Pseudoalteromonas, Marinobacter, Alteromonas, Pseudomonas, Oleispira, Oceanobacter, and Oceaniserpentilla (Supplementary Data 2). This indicates that viruses have potentially strong effects on the majority of deep-sea bacterioplankton. Alphaproteobacteria (e.g., SAR11 Clade Ia, Thalassococcus, Paracoccus) were the dominant groups in the in situ bacterioplankton community. In the +virus treatment, the abundance and activity of Alphaproteobacteria were repressed compared with the −virus treatment, resulting in a population shift to Gammaproteobacteria accounting for approximately 80% of total and active bacterioplankton. When viral pressure was relieved in the −virus treatment, Alphaproteobacteria dominating the original environments retained their dominant status. These bacteria may be comparatively inactive because of the differing conditions between the in situ and the microcosm environments. In the −virus treatment, Gammaproteobacteria and some Alphaproteobacteria (e.g., Sulfitobacter) could have outcompeted other bacterioplankton in terms of nutrient utilization or adaption to environmental changes, thus dominating the active bacterioplankton. Such a pattern, i.e., that numerically not dominating but potentially highly active bacteria are controlled by viruses, has been demonstrated for surface water29. In addition, the relative abundances of Bacteroidetes and Actinobacteria decreased after incubation, and the treatments with and without the pressure of virus lysis did not differ significantly, which suggest that these bacterial groups may be less susceptible to viral infection (Supplementary Fig. 3).
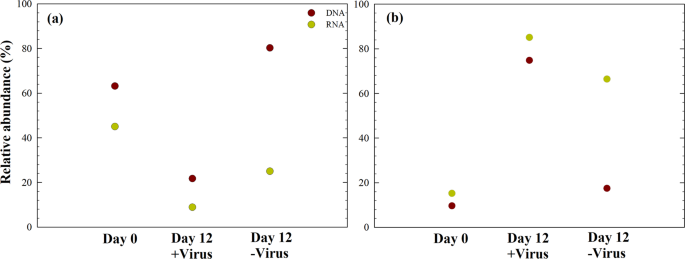
a Total and active Alphaproteobacteria, b Total and active Gammaproteobacteria. Data show the mean of two independent incubations.
Effects of viruses on the interactions among deep-sea bacteria
To evaluate whether the presence or absence of viruses may have affected the active interactions among deep-sea bacterioplankton during the 12-day incubation, we constructed co-occurrence networks for RNA-inferred bacterial communities (Fig. 7 and Supplementary Fig. 4; Table 1 and Supplementary Data 3), assuming that RNA rather than DNA reflects the active community. In network analysis, the topological indices mathematically describe the microbial interactions, including degree, number of nodes, density, and clustering coefficient30. The density, the proportion of actual connections to the potential connections in a network, is higher in the +virus treatment than in the −virus treatment. This indicates that more bacteria are connected and a more complex network when viruses were present (Table 1). Similarly, the higher clustering coefficient of +virus treatment than −virus treatment reveals the closer connection among the bacteria with the occurrence of virus (Table 1). The more complexity of microbial community shown by network density and clustering coefficient is consistent with the higher bacterial diversity found in the +virus treatments (Fig. 4). This could be explained by the concept of “kill the winner”24 that viruses control competitive dominates for nutrients and allow for the co-existence of more diverse bacterial community. Previous studies have shown that abiotic and biotic pressures might increase the network complexity of microbial communities31. Our data demonstrate that top-down pressure from viral lysis could have similar ecological influences. A positive correlation between two nodes in the microbial network is usually explained as a mutualism or cooperation relationship, while theoretically, this can also be produced by co-infection of different hosts by the same viruses. In our incubation, a lower proportion of positive correlations was observed in the +virus bacterial community compared with the −virus bacterial community (ca. 56% vs. 85%) (Table 1), suggesting that the positive correlation of bacteria may not be the direct result of bacterial mortality induced by viral lysis. This is reasonable as viral infection is always host-specific and their host range is narrow. Also, viral lysis does not only cause mortality of hosts (hence influence competition among bacteria), but also causes a release of the cell content and cell wall content (lysis products) which can be used by bacterioplankton (“viral shunt”). This supposedly “lubricates” the microbial food web and could result in positive correlations of the network analysis.
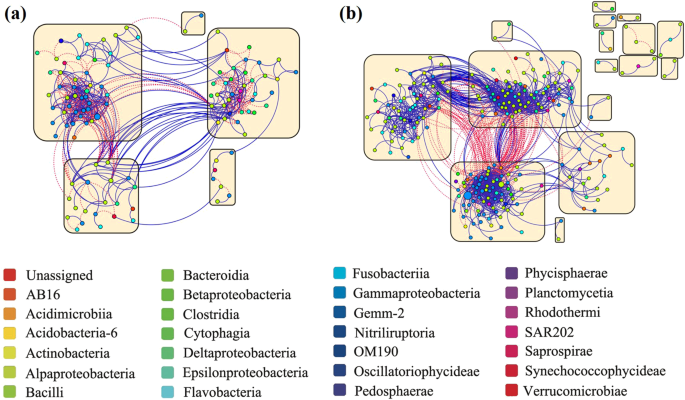
a Co-occurrence network graph of bacterioplankton with normal viral lysis pressure. b Co-occurrence network graph of bacterioplankton with reduced viral lysis pressure. The nodes, representing OTUs, are colored according to phylogenetic taxa. Node size is proportional to the relative abundance of OTU. Blue and red lines indicate positive and negative connections, respectively, between nodes. Yellow squares show modules in the network.
Four large modules contained more than five nodes were observed in each of +virus and −virus treatments, showing an evident modular architecture for both networks (Table 1). Since the OTUs within a module might occupy a similar ecological niche, a higher modularity value and module number observed in the −virus treatment (Fig. 7) indicated that number of niches increased without virus. In addition, the presence of virus stimulated the connections among bacteria within the module, which was evidenced by the higher clustering coefficients and increased proportion of links within modules in the +virus treatment (Table 1 and Fig. 7). Thus, the presence of virus might facilitate the stability of the niches by increasing the interactions among the bacteria in each niche. We also measured the proportions of Alphaproteobacteria and Gammaproteobacteria (the two major bacterial groups in the present study) in the networks (Supplementary Data 3). The Alphaproteobacteria to Gammaproteobacteria ratio was higher in the +virus treatments than in the −virus treatments, and the proportion of positive correlations for Gammaproteobacteria was greater than that for Alphaproteobacteria. Our results suggest that the removal of viruses might change the connection type (positive or negative) among bacteria, especially the Gammaproteobacteria. Thus, viruses could play a fundamental role in sustaining the deep-sea ecosystem by shaping the interactions among bacteria.
Methodological considerations
In our study, we used onboard microcosm incubation (20 L) in the dark at the in situ temperature to represent deep-sea environments. A main difference between our incubation and the in situ deep-sea environment was pressure. The inactive deep-sea microbial community (e.g., allochthonous and piezosensitive) may become dominant while piezophilic and hyperpiezophilic microorganisms may disappear when incubated under atmospheric pressure32. However, there are evidences that pressure did not significantly affect viral activities and, subsequently, the impact of viruses on their host population13,33. In addition, the enclosure of microbial plankton communities may be biased by “bottle effects”, which may either stimulate the growth or enhance the loss of different planktonic groups34,35. However, if we assume that the “bottle effects” and effects of pressure changes in the +virus and −virus treatments were similar (as all incubation conditions were the same), differences in the ecological characteristics of bacterioplankton (Figs. 2–7) in our study should be due to the presence or absence of the top-down pressure of viral lysis. For example, the replacement of Alphaproteobacteria by Gammaproteobacteria has been frequently observed in microcosm incubations of bacterioplankton, and this was usually explained as resulting from the fast growth rate and opportunistic life strategy of Gammaproteobacteria in bottle incubations36,37. In our study, however, the simultaneous incubation in the −virus treatment showed a contrasting pattern: Alphaproteobacteria predominated in the DNA library, while Gammaproteobacteria predominated in the RNA library. This difference clearly indicates that viruses can also play a major role in shaping the bacterial community structure in the deep sea. In addition, the PCR-based method was applied to estimate bacterial diversity and relative abundance. Although we controlled the PCR cycle numbers and performed triplicate amplification for each sample to minimize any possible bias introduced by PCR, the relative abundance estimation may be biased by the variation of 16S rRNA gene copy number in the deep-sea bacterial genome (e.g., see ref. 38).
Source: Ecology - nature.com
