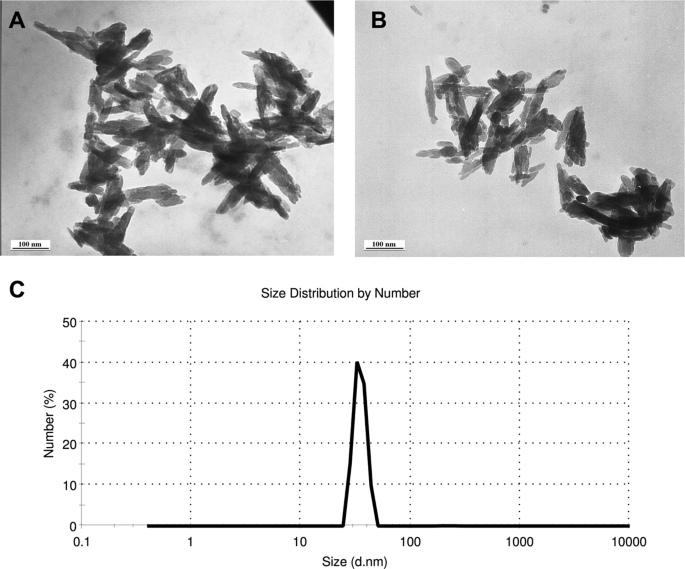
Nanoparticle characterization
Although nano-fertilizers are advertised for sale to growers from several companies in China and India, full details of their compositions were not available. Thus, characterized hydroxyapatite (Ca10 (PO4)6(OH) 2) nanoparticles (nHA) were purchased from Sky Springs Nanomaterials, Inc. (Houston, Texas, USA) as <40 nm, with a needle shaped morphology, and 98.5% purity. nHA shape and size was confirmed using transmission electron microscopy (TEM) after suspension in Milli-Q water and sonication for 10 min, with mean nHA length determined using ImageJ33. Sonicated Milli-Q water suspensions of nHA were used to verify particle size distribution and average size by DLS analysis using a Malvern Zetasizer Nano ZS.
nHA microbiome experiment
Farm soil mixture
Peat-pearlite SunGro Sunshine® Mix #2 soil with no added fertilizer (soluble Pi < 10 µg/g34) was sifted to remove large twigs and autoclaved twice to ensure sterility. It was then inoculated with field soil (5% w/w) obtained from a farm north of Sydenham, ON (44.4°N, 76.6°W) which had not been treated with fertilizer for 25 years. The soils were thoroughly mixed using a cement mixer for 15 min, bagged, and incubated at 22–25 °C until further use.
Experimental treatments and cultivation
A total of 140 pots (13 cm) were used, consisting of treatments with 20 pots each sampled for microbiomes (n = 100), 5 replicate pots used for monitoring growth under experimental conditions (n = 25), and three extra replicates per treatment (Table 1). Additional +P control pots fertilized with soluble Pi were monitored separately (Table S1, see Supplementary Information).
Each pot contained 86 g of the soils (95:5 sterile potting soil and farm soil, respectively, and incubated to “seed” the mixture with field-derived microorganisms). HA and nHA additions were made at the concentrations indicated (treatments C-E; Table 1) in a depression (3 cm in diameter and 1 cm deep) made in the soil mixture (Fig. S4A). Three soybean seeds (Wallace Variety, Willow Agriservice, Harrowsmith, ON) were planted 1 cm below the soil surface around the centre of the pot (Fig. S4A). The seeds had been previously coated with HiStick® N/T Soybean co-inoculant (containing Bradyrhizobium japonicus and Bacillus subtilis MBI600) at the recommended rate (3.2 mg/g of seed). After covering seeds and nHA/HA with soil, pots were watered with reverse-osmosis (RO) water (200 mL, as determined empirically). Pots were placed in a growth chamber set to 23–24 °C during the day, 20 °C at night, with a photoperiod (estimated at 600 mE/m2s1) of 13 h35. Plants were rotated within the chamber every other day to minimize place effects. One week after planting, the seedlings were thinned so that one plant remained per pot. Treatment groups B-E were fertilized with 50 mL of 3.5 mM KNO3 solution to facilitate the formation of nodules for the first week after planting22, and subsequently fertilized every other day with 50 mL of Hoagland’s solution lacking nitrogen and phosphorus (see Supplementary Methods, Table S12)36. Treatment A pots were provided with a comparable 50 mL of RO water daily, with all plants in each treatment group watered every other day with RO water as required.
The 5 growth replicates per treatment were cultivated for 8 weeks and fertilized as described. Above ground and below ground plant tissues were then collected, dried (48 h at 70 °C), and weighed. Pods and root nodules were counted. Nodules were dissected to determine reddish-pink colouring, an indication of active nitrogen fixation37.
Sample collection
After three weeks, all plants had reached the V2 stage (2nd trifoliate stage) and samples from the leaf, root, and soil were collected for microbiome analysis. From each of the 20 plants per treatment the middle leaf of the first trifoliate was removed with ethanol-sterilized forceps and kept at 4 °C for ~2 h in a sterile bag until processed. Four cores were then aseptically removed from different areas of each leaf using a flame-sterilized cork borer (1 cm diameter), placed in a 1.5 mL centrifuge tube, snap frozen with liquid nitrogen, stored at −80 °C, and designated as the endosphere/phyllosphere (above ground on and within plant microbiome).
Soil samples were collected using a scoopula sterilized with 70% ethanol from the center of each pot, and after removing the top 1 mm of soil, enough soil was collected from each pot to fill a 15 mL conical centrifuge tube. Samples were stored at −20 °C until they were lyophilized prior to extraction. Roots were gently and aseptically separated from the remaining soil and stored at 4 °C in sterile bags overnight. A 2.5 cm root section was cut with sterilized scissors 2.5 cm down from the stem-root junction, and subsequently any adhering nodules and lateral roots were removed from the root section samples. The root samples were placed in 1.5 mL centrifuge tubes containing 1.3 mL of Pi-buffered saline (PBS) containing 1 mM EDTA and 0.01% Tween-20, and shaken for 20 min at 180 rpm, vortexed for 15 sec and then transferred into a new tube. The remaining root wash solution, containing any microbial cells and DNA washed from root surface, was centrifuged (20 min at 335 × g) and the supernatant removed, with the remaining pellet frozen in liquid nitrogen and stored at −80 °C and designated as the rhizosphere sample.
DNA extraction and electrophoresis
Soil and rhizosphere DNA samples were extracted using the NucleoSpin® Soil Genomic DNA Extraction Kit (Macherey-Nagel) following the manufacturer’s protocol but with the addition of a repeated sample lysis step (SL1 lysis buffer and Enhancer SX). For soil samples (150 mg), 150 μL of extra lysis buffer was used. Pelleted rhizosphere material (50–290 mg; mean 125 mg) was initially suspended in SL1 lysis buffer and transferred to bead tubes with the rest of the extraction carried out following the manufacturer’s protocol.
Phyllosphere/endosphere DNA was extracted with the Wizard® Genomic DNA Purification Kit (Promega, WI, USA) following the manufacturer’s protocol for the isolation of plant genomic DNA, with an added lysozyme step38. All DNA extractions were quantified using both Nano-Drop and Qubit® Fluorometer (Invitrogen, ThermoFisher). DNA quality was also verified using 1% agarose gel electrophoresis with samples stored at −20 °C before further analysis. Prior to sequencing, community structure of the bulk soil was examined using PCR-denaturing gradient gel electrophoresis to verify the presence of a detectable bacterial community (not shown).
rRNA and ITS gene amplicon sequencing and analysis
For each of the 5 treatments from the bulk soil, rhizosphere, and phyllosphere/endosphere samples, 10 out of the 20 replicate DNA samples from each treatment were randomly selected for 16S rRNA and ITS gene amplicon sequencing (150 total). DNA samples were sent to the Centre for the Analysis of Genome Evolution & Function (CAGEF) at the University of Toronto for sequencing. The V4 hypervariable region of the 16S rRNA bacterial gene fragments were amplified using a universal forward sequencing primer (515 F) and a uniquely barcoded reverse sequencing primer (806 R) to allow for multiplexing39. Amplification reactions for the soil and rhizosphere samples were performed using 12.5 μL of KAPA2G Robust HotStart ReadyMix (KAPA Biosystems), 1.5 μL of 10 μM forward and reverse primers, 8.5 μL of sterile water and 2 μL of DNA. The V4 region was amplified by cycling the reaction at 95 °C for 3 min, 18x cycles of 95 °C for 15 sec, 50 °C for 15 sec and 72 °C for 15 sec, followed by a 5 min 72 °C extension. For the peptide nucleic acid (PNA) clamp reactions using leaf DNA extract as the template, 2 μL of DNA was added to 12.5 μL of KAPA2G Robust HotStart ReadyMix (KAPA Biosystems), 1.5 μL of 10 μM forward and reverse primers, 6 μL of sterile water, 0.75 μL of 25 μM plastid PNA clamps and 0.75 μL of 25 μM mitochondrial PNA clamps (PNA BIO Inc., Newbury Park, CA). The V4 region was amplified by cycling the reaction at 95 °C for 3 min, 20x cycles of 95 °C for 15 sec, 78 °C for 15 sec, 50 °C for 15 sec and 72 °C for 15 sec, followed by a 5 min 72 °C extension. The fungal ITS1(internal transcribed spacer) region was amplified using ITS1F and ITS1R primer sets40 as described for the V4 procedure but by reducing the DNA template to 1 μL and optimizing amplification with 25 cycles of 95 °C for 15 sec, 56 °C for 15 sec and 72 °C for 15 seconds, followed by a 5 min 72 °C extension.
For both bacterial and fungal DNAs, all amplification reactions were done in triplicate as well as negative controls for each barcode, verified on a 1% agarose TBE gel, and then pooled to reduce amplification bias. Pooled triplicates were combined by approximately even concentrations as determined by a Qubit fluorometer. Standard Nextera XT protocols were followed, selecting for 300–500 bp fragments, with the final libraries purified using 0.8X magnetic Ampure XP beads, again quantified using fluorescence (Qubit) and each of the bacterial and fungal libraries were separately pooled and sequenced, according to manufacturer’s instructions (Illumina MiSeq, San Diego, CA). For both library sets, sequencing was performed using V2 (150 bp × 2) chemistry
The UNOISE pipeline, available through USEARCH version 9.2, was used for sequence analysis41,42,43. The last base, which is typically error-prone, was removed from all the sequences. Sequences were assembled and quality trimmed using –fastq_mergepairs and –fastq_filter set at 1.0, with a –fastq_maxee set at 0.5. Sequences less than 233 bp (20 bp shorter than the average) were also removed. For fungal ITS analysis, following the UNOISE pipeline, unique sequences were identified from the merged pairs and for all analysis, merged pairs were de-replicated and sorted to remove singletons. Sequences were denoised and chimeras were removed using the unoise2 command. For 16S rRNA gene analysis, assembled sequences were mapped back to the chimera-free denoised sequences at 97% identity OTUs. Taxonomy assignment was executed using utax and the UNOISE compatible Ribosomal Database Project (RDP) database version 16, available to download through USEARCH, with a minimum confidence cutoff of 0.944. OTU sequences were aligned using PyNast accessed through QIIME45. Sequences that did not align were removed from the dataset and a phylogenetic tree of the filtered aligned sequence data was made using FastTree46. For ITS analysis, assembled sequences were mapped back to the chimera-free denoised sequences at 97% identity OTUs using the –otutab command. Taxonomy assignment was executed using SINTAX43, available through USEARCH, and the SINTAX compatible Ribosomal Database Project (RDP) Warcup ITS v2 database, with the default minimum confidence cut-off of 0.844.
Prior to analysis, both bacterial and fungal OTU tables were filtered to remove any OTUs represented by sequences numbering less than 0.005% of the total47. Any unassigned OTUs were also removed prior to analysis. The OTU tables were separated based on sample type to create individual OTU tables for soil, rhizosphere, and leaf samples (endosphere/phyllosphere), as applicable, which were then analyzed separately. The majority of the subsequent OTU table analysis was done using QIIME (1.9.1)45, including data normalization, taxonomic relative abundance, rarefaction, alpha diversity calculation, and beta diversity calculations. Alpha diversity was calculated on data rarefied to the minimum sample counts using both Faith’s phylogenetic diversity index (PD) and the Shannon index for 16S rRNA gene analysis, while observed species (OS) and Shannon index were used for fungal ITS data. Prior to beta diversity analysis, data were normalized using both rarefaction to the lowest sample count and also cumulative sum scaling (CSS) normalization21. Beta diversity was calculated using the weighted UniFrac metric, which takes into account the taxonomic relationship and abundance of OTUs within samples48. Principle coordinates analysis (PCoA) and generation of heatmaps were done using QIIME.
Statistical analysis
Statistical analysis of plant biomass and pod numbers was done using one-way ANOVA and TukeyHSD post-hoc tests calculated through GraphPad Prism (version 7.0, GraphPad Software, La Jolla, CA, USA). Non-parametric ANOVA (Kruskal-Wallis test) run through R (version 3.3.2) was used to determine significant differences in phylum and genus relative abundance between treatments, with post-hoc testing done using Dunn’s multiple comparisons test implemented through the FSA package in R. Differences in alpha diversity were also tested using non-parametric ANOVA, here implemented through GraphPad Prism. Analysis of the statistical significance of sample groupings for beta diversity metrics was done using the compare_categories.py script in QIIME, which uses the R vegan package to run both ANOSIM and adonis (PERMANOVA) tests using 1000 permutations. Finally, statistical analysis of beta diversity distances within and between treatments was done using the make_distance_boxplots.py script in QIIME, which ran parametric t-tests with Bonferroni P-value correction for multiple comparisons. All statistical tests were conducted using a significance level (α) of 0.05.
Growth and production experiments
Experiment set-up
Six treatments (A-F) were set up in greenhouse experiments (Table 3). Dry peat-pearlite (500 g) SunGro Sunshine® Mix #2 (contains no added fertilizer, and not autoclaved as microbiomes were not examined) was placed into 100 pots (20 cm diameter) and then wetted with RO water. Additions were made to the pots as required (treatments C-E) in the centre of each pot. A 50 mL conical centrifuge tube was used to lift a core of soil 7.6 cm deep, below which the HA or nHA was placed before replacing the overlaying soil. Soybean seeds (3) as previously described, were planted 2.5 cm below the surface surrounding the centre of all pots (Fig. S1B) and watered. Two weeks after planting, seedlings were thinned to one per pot, with un-germinated seeds also removed from the pots. Notably, in these experiments, soybean seeds were surface sterilized by successively immersing them for 1 min in 70% ethanol, sterile water, 10% sodium hypochlorite, sterile water and subsequently rinsed 3X with sterile water before potting. Surface sterilization was intended to remove or kill fungi or bacteria, including nitrogen-fixing bacteria, as experimental treatments supplied the nitrogen. For treatment groups C-E (Table 3), HA or nHA was placed 5 cm below the soybean seeds in the centre of the pot in accordance with agricultural recommendation as well as fertilizer concentrations as suggested for soybean (Ontario Ministry of Agriculture, Food and Rural Affairs; OMAFRA). Levels of 20 kg/ha P2O5 represent the lowest recommended field application rate when the soil test level indicates a P concentration of 13–15 ppm49. Thus nHA/HA was added in treatment E to provide equimolar amounts of P in the form of Ca10(PO4)6(OH)2 dictated by the recommended application rate of 20 kg/ha P2O5 based on the surface area of the 20 cm pots used for this experiment. The lowest concentration of P in the soil recommended to grow soy is 15 ppm, which is considered the critical P concentration below which crops experience decreased yield due to P-deficiencies50. This concentration of 15 ppm P in the form of HA or nHA was used in treatment C and D respectively, based on the dry mass of soil used in the pots. This concentration was informed by the previous reports indicating that nHA was more effective than soluble P fertilizers (P2O5) at the same rate of application8.
Soy was grown in a greenhouse (20–24 °C with a photoperiod of 18 h). The first replicate experiment (GP1) was conducted for 12 weeks (late April-July), with a second replicate experiment (GP2) similarly conducted (mid-May- late July). Two weeks after planting and approximately one week after germination, 350 mL Plant-Prod® 14-0-14 Balance Fertilizing solution was applied weekly to treatments B-E (Table 3) at a rate of 100 ppm N with 350 mL of RO water given weekly to treatment A. Treatment F was given 350 mL of Plant-Prod® 20-20-20 weekly also at a rate of 100 ppm N. Additional water was applied equally to all treatments as required. Soy growth was monitored weekly for the duration of the experiments. Height was recorded weekly and the number of trifoliate leaves, seed pods, and amount of foliage lost was recorded for each plant.
During GP1 replicate growth, thrips were observed in the greenhouse, resulting in mild foliar damage 10 days after planting. Subsequently, predatory Swirski-Mites (Koppert Biological Systems) biocontrols were applied once every fortnight to each plant and thrip presence was monitored as recommended by a modified version of Purdue University’s protocol51. Briefly, each of the four greenhouse benches was demarcated in 5 zones, from which a trifoliate leaf (GP1: uppermost trifoliate; GP2: lowest trifoliate) from 5 randomly selected plants were enumerated for the presence of thrips at any life stage.
Sample collection for biomass and yield
After 12 weeks of growth, all GP1 and 2 plants were harvested. The above ground portion of the plant was cut from the roots ~1 cm above the first lateral root and placed in a paper bag for drying. Roots were carefully separated from the soil, washed with water and bagged as previously described. They were then dried for 2 days at 70 °C. Subsequently, pods were removed and weighed separately, after which all seeds were removed from the pods and placed back in the drying oven for an additional 2 days. Total dry weight of the roots and the above ground tissue (without seeds) was used as an estimate of soy plant biomass. Dry mass of seeds was recorded, as a measure of seed yield.
After data recording, 9 random samples from each treatment were used to determine above ground total plant P concentrations. Oven dried samples were pooled in triplicate and ground using a coffee grinder to prepare samples for analysis. Total plant tissue P concentrations were determined using ICP-OES (inductively coupled plasma-optical emission spectroscopy), which was conducted by the Analytical Services Unit (ASU) at Queen’s University. Spinach (Spinacia oleracea) leaf tissue was used as an analytical reference, along with a separate P control sample. Above ground plant tissues analyzed included the stem, remaining leaves, and empty pods. Analysis of soluble Pi in soil samples collected from three random pots for treatments A-E was also done using ICP-OES. Prior to analysis, 0.5 g of each dried soil sample (oven dried at 70 °C for 2 days) was added to a glass vial with 40 mL of water. Sample vials were then placed on an orbital shaker overnight at 300 rpm to extract any soluble Pi. After shaking, the sample mixture was syringe-filtered (0.45 μm). Filtered solutions were then analyzed to determine Pi concentration using ICP-OES with a standard Pi control (ASU). For the plant growth and production experiments, as described earlier, statistical analysis was performed using R Studio (R version 3.3.2) and GraphPad Prism (version 7.0). One-way ANOVA with TukeyHSD post-hoc tests were used to determine any statistically significant differences between groups for final height, trifoliates produced, overall biomass, below and above ground biomass, and seed yield all using a significance level of 0.05.
Accession codes
Biological sequencing data is available from NCBI Sequence Read Archive under the BioProject accession number: PRJNA544311.
Source: Ecology - nature.com
