Transcriptome sequencing and de novo assembly
RNA sequencing (RNA-seq) experiment was performed on 6 samples: a triplicate derived from the microalga C. closterium cultured in complete medium and considered as control condition (CTRL), and a triplicate of C. closterium cultured in Si-starvation conditions (Si-starved). Since the genome of C. closterium is not available, RNA-seq reads have been assembled with de novo approach producing 44718 putative transcripts grouped into 33433 genes. The mean GC content was 46.16%. The average and the median contig length were 1064 bp and 781 bp, respectively. The N50 was 1554 bp.
Several controls were performed on the raw transcriptome assembly to check for its quality. Transcripts were translated into proteins obtaining a total of 32586 protein sequences (minimum length 50aa). Among those, 14307 (43.90%) were complete (with a methionine and a stop codon), 2855 (8.76%) started with a methionine but lacked a stop codon, 8194 (25.14%) only had a stop codon, 7230 (22.18%) did not start with a methionine and did not have a stop codon.
In order to verify the completeness of the assembly, the protein sequences were blasted against the two datasets of the Core Eukaryotic Genes, including 248 (http://korflab.ucdavis.edu/Datasets/genome_completeness/) and 458 (http://korflab.ucdavis.edu/datasets/cegma/) protein sequences, respectively. 248 out of 248 (100%) and 456 out of 458 (99.56%) could be detected in the assembly. In addition, the length of the proteins from the assembly was compared to the length of the core eukaryotic genes. About 403 of the proteins covered more than 90% of the length of the corresponding core eukaryotic proteins and 536 covered more than 80% of the length of the corresponding core eukaryotic protein, indicating a good quality of the assembly.
As NGS data could suffer from contaminations of organisms that are not the target of the experiment, we blasted the sequences of the transcripts against the NCBI database of bacteria and archaea in order to remove possible contaminations. By this way, 172 transcripts were detected and removed. In order to detect other sources of contamination, the distribution of the GC content was analysed in the dataset. As already mentioned, the GC content followed a normal distribution with a mean value of 45.84% and a standard deviation of 3.48. Following a z‐test, 5445 sequences could be identified for having a GC content significantly different from the observed mean (p < 0.01). Those sequences were blasted against the NCBI (nr) database to look for contaminants. About 878 sequences were removed for matching bacterial or metazoan sequences.
The obtained assembly was initially composed by 43668 transcripts belonging to 32606 genes. The mean GC content was 46.11%. The average and the median contig length were 1079.06 bp and 803 bp, respectively. The N50 was 1564 bp.
Functional annotation
Assembled transcript sequences were translated into proteins with Transdecoder (minimum length of 50 aa). When multiple translations were possible, the priority was set in order to get the longest complete ORF; when a complete ORF was not detected the longest sequence was kept. The sequences were also investigated for the presence of repetitive elements with Repeat Masker. The software Blast2GO was used to associate a function to the assembled transcripts. A total of 20540 proteins got significant blast hits, and among these 11178 proteins had gene ontology (GO) terms associated. During the blast step, we realized that some proteins had human or bacterial hits, 355 and 103 respectively. These sequences were removed from downstream analysis. Finally, the filtered assembly was composed by 43210 transcripts grouped into 32251 genes (Table 1). The mean GC content was 46.10%. The average and the median contig length were 1078.34 bp and 803 bp, respectively. The N50 was 1561 bp. The final dataset was then translated into proteins (minimum length 50 aa) obtaining a total of 31613 protein sequences. Among these, 14162 (44.79%) were complete protein sequences (with a methionine and a stop codon), 2782 (8.8%) started with a methionine but lacked a stop codon, 7957 (22%) had only a stop codon, 6712 (25.17%) did not start with a methionine and did not have a stop codon.
Differential expression analysis
Differential expression analysis identified 1818 genes with significant expression variations in Si-starved condition compared to control. Among them, 845 were up-regulated (among these, 191 had an NCBI NR assignment), while 973 were down-regulated (among these, 361 had an NCBI NR assignment). The full list of DEGs, log fold change (logFC), false discovery rate (FDR) and their NCBI NR assignment are reported in the Supplementary Table 1.
Among the Differentially Expressed Genes (DEGs), Si-starvation induced an up-regulation on various unknown proteins, tkl dicty4 protein kinase (FDR < 1,35E-004), rna pseudouridine (FDR < 8,82E-003) and a transketolase (FDR < 0). Conversely, Si-starvation induced a strong down-regulation of vacuolar iron family transporter (FDR < 0), sodium bile acid cotransporter 7 isoform x1 (FDR < 6,48E-005) and two ABC transporters (FDR < 6,97E-009 and 1,01E-003, respectively).
Up-regulated transcripts were mainly involved in pathways related to bicarbonate transporters (R-HSA-425381), integrin cell surface interactions (R-HSA-216083) and unwinding of DNA (R-HSA-176974). On the contrary, transcripts involved in pathways related to Na+/Pi cotransporters (R-HSA-427589), transport of bile salts, organic acids, metal ions and amine compounds (R-HSA-425366), ABC transporters in lipid homeostasis (R-HSA-1369062), biotin transport (R-HSA-196780) and metabolism of folate and pterines (R-HSA-196757) were down-regulated when cultured in Si- starvation. In order to validate differential expression analysis between the control and Si-starvation conditions, 13 transcripts were tested using reverse transcription-quantitative PCR (RT-qPCR; gene names and primers are reported in Supplementary Tables 2 and 3). A significant positive correlation was established between RNAseq and RT-qPCR analyses (R = 0.8767, p value < 0.00001), supporting the results obtained by RNA-Seq.
Identification of transcripts related to anti-inflammatory compound metabolism
We identified 5 transcripts coding for enzymes involved in the synthesis of compounds with possible anti-inflammatory activity: Phosphatidylinositol-3-Phosphatase (PtdIns(3)), Phosphatidylinositol 3-kinase catalytic subunit type 3 (PIK3C3), Phosphatidylinositol N-acetylglucosaminyltransferase subunit A (PIGA), Monogalactosyldiacylglycerol synthase (MGD), and Violaxanthin De-Epoxidase (VDE). These were selected as genes of interest (GOI) for this study.
Several inositol derivatives, such as phosphatidylinositol, are known to have anti-inflammatory activity26,30,31 and have been also quantified in several microalgae to be proposed as supplement in diet32. PtdIns(3), PIK3C3 and PIGA are the main enzymes involved in the inositol phosphate metabolism (KEGG pathway ec00562), and were selected as GOI for RT-qPCR analyses in this study. In particular, PtdIns(3) converts 1-phosphatidyl-1D-myo-inositol 3-phosphate in 1-phosphatidyl-1D-myo-inositol, while PIK3C3 and PIGA convert a 1, 2-diacyl-sn-glycero-3-phospho-(1D-myo-inositol) in a 1, 2-diacyl-sn-glycero-3-phospho-(1D-myo-inositol 3-phosphate) and a 6-(N-acetyl-α-D-glucosaminyl)-1-phosphatidyl-1D-myo-inositol, respectively (https://www.uniprot.org/uniprot/Q6PF93, https://www.uniprot.org/uniprot/P37287). Phosphatidylinositol 3-kinase is known to regulate key events in inflammatory responses, and there are also studies on genetically-modified mice with altered phosphatidylinositol 3-kinase signaling in order to understand its role in chronic inflammation mouse models33. Another selected transcript was MGD, essential for the synthesis of monogalactosyldiacylglycerols (MGDG)34. Several microalgae are known to be rich in MGDG containing a high proportion of polyunsaturated fatty acids (PUFAs) with potential nutraceutical and pharmaceutical applications35. MGDG are known to have anti-inflammatory activity reducing the release of inflammatory mediators (i.e. interleukins) and inhibiting the generation of superoxide anion34.
Finally, the transcript coding VDE was selected as well. VDE converts violaxanthin to zeaxanthin and both of them, as well as other carotenoids, are known to possess anti-inflammatory activities36,37 and have been widely studied in microalgae for their potential health benefits15. Primers were designed in order to amplify the five selected transcripts, their expression levels were analyzed in both control and Si-starvation conditions by RT-qPCR and in silico prediction of the three-dimensional structures of their corresponding proteins was performed.
Reference gene assessment and GOI expression level analyses
In order to study GOIs expression levels, a panel of putative reference genes (RGs) was screened. Selected genes were included in those already used as RGs for other microalgae38,39,40,41,42. Results on the assessment of the best RGs obtained by three different software are reported in Fig. 1. According to the results obtained by BestKeeper, the lowest standard deviation (SD) was obtained for α-tubulin (Tub-α), followed by calmodulin (CaM) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Fig. 1a), which indicated that the most stable RG was Tub-α. According to NormFinder, the lowest stability values were calculated for GAPDH, Act and CaM; therefore, they were suggested as best candidate RGs (Fig. 1b). According to the statistical approach of geNorm, the two most stable genes (i.e., with the lowest expression stability, M) were actin (Act) and translation elongation factor-like protein (EFL) (Fig. 1c). Pairwise variation was calculated for assessment of the effect of adding another RG to those already analysed. The obtained results indicated that the addition of other RGs was not required since the value for V2/3 is below the cutoff of 0.15 (Fig. 1d). Considering the best RGs assigned by each software, Tub-α, GAPDH, Act and EFL were selected as RGs for RT-qPCR analyses. The best predicted RGs were different compared to other diatoms (such as Skeletonema marinoi, Pseudo-nitzschia multistriata and P. arenysensis39,40,42 since their stability is related to the studied species, growth phases and studied conditions (e.g. nutrient starvation/depletion and CO2 exposure).
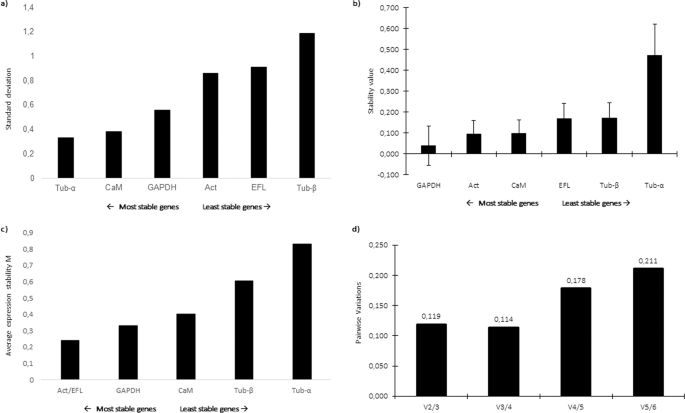
Reference gene assessment for Cylindrotheca closterium. Ranking of the best reference genes obtained with (a) BestKeeper (lowest standard deviation), (b) NormFinder (lowest expression stability value) and (c) geNorm (lowest average expression stability M) softwares. (d) According to geNorm algorithm, the inclusion of additional reference genes was not required for values below the cut-off of 0.15. The selected genes were: Actin (Act), Glyceraldehyde 3-Phosphate Dehydrogenase (GAPDH), Translation elongation factor-like protein (EFL), Calmodulin (CaM), α-tubulin (Tub-α) and β-tubulin (Tub-β).
Relative expression levels of the selected GOIs were analyzed in order to investigate the effect of Si-starvation on their expression. Figure 2 displays GOI expression levels in C. closterium cultured in Si-starvation compared to the control condition (x-axis). According to REST analysis, among the GOI, Gal-9, PIGA, PK3C3 and VDE showed significant down-regulation under Si-starvation (p < 0.001 for all), suggesting that there is higher probability to find their corresponding proteins and products in control conditions. For the other transcripts, changes were not significant. Even if whole transcriptome expression analyses in response to Si-starvation were already done for Phaeodactylum tricornutum43 and Thalassiosira pseudonana44,45, our GOIs were not previously studied. Hook et al.46 also sequenced the transcriptome of a C. closterium clone using 454 pyrosequencing and investigated its ecotoxicogenomic response by exposing the microalgae to various coastal contaminants (e.g. a photosystem II inhibiting herbicide, ammonia, copper and crude oil). However, due to differences in the aims of the studies and in the studied genes, a direct comparison with the present work was not possible.
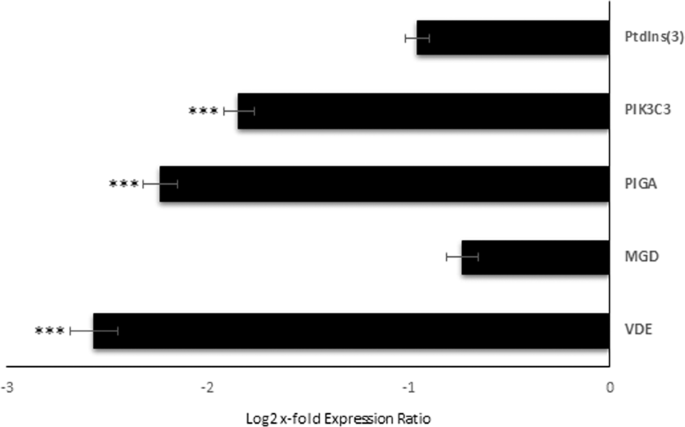
Expression levels of selected genes of interest in C. closterium cells cultured in silica starvation compared to the control condition. Data are represented as log2 x-fold expression ratio ± SD (n = 3). C. closterium cultured in complete medium was used as control (represented in the figure by x-axis; *** for p < 0.001). Gene abbreviations were Phosphatidylinositol-3-Phosphatase (PtdIns(3)), Phosphatidylinositol 3-kinase catalytic subunit type 3 (PIK3C3), Phosphatidylinositol N-acetylglucosaminyltransferase subunit A (PIGA), Monogalactosyldiacylglycerol synthase (MGD), and Violaxanthin De-Epoxidase (VDE).
Domain identification and structure prediction of proteins encoded by selected GOI
With the aim of supporting the quality assessment of the selected GOIs, we performed an InterProScan analysis on the entire protein collection of C. closterium, obtaining an exhaustive functional characterization for each protein sequence. In Table 2, functional annotations from PFAM (protein families) and InterPro (protein domains) databases of five selected GOIs involved in anti-inflammatory compound metabolism are shown. This is the first study to investigate and identify transcripts involved in the synthesis of inflammatory mediators in diatoms and in silico predicting their three-dimensional structures.
In order to further investigate the selected GOIs at protein structure levels, we in silico predicted their three-dimensional structure by fold recognition approach (Kelley et al. 2015 https://www.ncbi.nlm.nih.gov/pubmed/25950237) (Fig. 3). The modeled structures showed PHYRE2 confidence scores that range between 99, 5% and 100%.
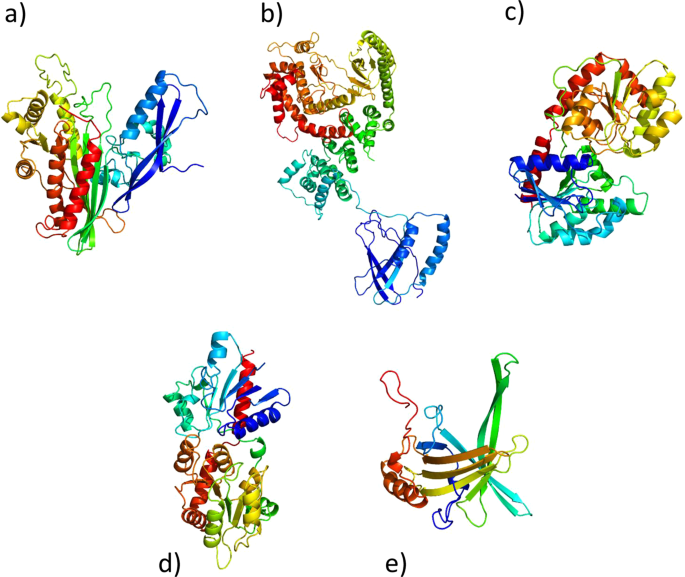
In silico protein structure predictions of five proteins encoded by the selected genes of interest. Protein structure predictions, colored by rainbow from N to C terminus, for Gal-9 (a), PtdIns(3) (b), PIK3C3 (c), PIGA (d), MGD (e) and VDE (f). Three-dimensional structures are indicated as follows: α-helices are represented by helices, β-strands are represented by arrows, coils are represented by simple lines.
Anti-inflammatory testing
C. closterium anti-inflammatory activity was screened by monitoring the release of tumor necrosis factor α (TNFα) in human THP-1 cells. Microalgae cultured in Si-starvation condition did not show anti-inflammatory activity (8% TNFα-inhibition, p > 0.05). Similarly, previous studies have shown that C. closterium cultured in nitrogen- and phosphate-starvation did not show anti-inflammatory activity, while the same species cultured in control condition (complete medium) induced more than 80% TNFα-inhibition10. These data suggest that in stressful conditions, such as nutrient starvation, C. closterium should not produce anti-inflammatory molecule/s or produce very low amounts. This information will direct future chemical analyses in C. closterium cultured in control conditions and guide marine natural product discovery increasing diatom applications in the blue biotechnology sector.
Source: Ecology - nature.com
