Phylogeny of Rhodocyclales based on 16S rRNA gene
Consistent with the existing classification result only based on type strains14, phylogenetic analysis on the basis of 16S rRNA gene sequences supported that the order consisted of four family-level lineages, namely, Rhodocyclaceae, Azonexaceae, Zoogloeaceae and Azovibrio_f (Figs. 1 and S2). Zoogloeaceae exhibited a considerably higher intra-lineage diversity and phylogenetic depth than the other lineages. Rhodocyclaceae and Azonexaceae were moderately supported with bootstrapping values over 50% for at least three methods (ML, NJ and BY). However, the two other lineages were only weakly supported by one or two methods. As expected, this result based on the rRNA gene provided an ambiguous classification, as pointed out by previous studies52,53. By contrast, the genus-level phylogeny was consistent with the current taxonomic system in most cases, except for several sequences (not from type strains) that were previously defined as Dechloromonas and Azospira.
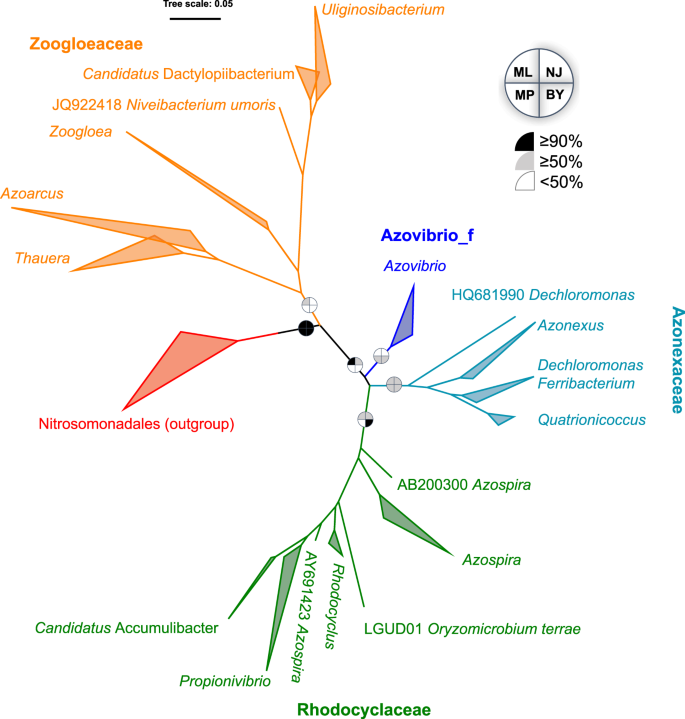
Maximum-likelihood tree on the basis of the 16S rRNA gene of Rhodocyclales. The bootstrap values on nodes indicating the percentage of reconstructions in which the topology was preserved using the maximum-likelihood (ML), neighbor-joining (NJ), maximum-parsimony (MP) and Bayesian (BY) methods. Node values are Bayesian posterior probability support values. Scale bar represents substitutions/site. Colored branches and labels indicate major clades. The outgroup contained sequences from Nitrosomonadales.
Five family-level sub-lineages in Rhodocyclacles indicated by phylogenomics
As shown in Fig. 2, the phylogenomic tree contained 90 predefined Rhodocyclales genomes and two genomes from Nitrosomonadales as outgroups (Sulfuritalea hydrogenivorans and Methyloversatilis discipulorum, which were previously incorrectly assigned in Rhodocyclales13). It was very clear that this phylogenomic tree had a higher resolution and provided more robust and accurate phylogenetic relationships (Fig. 2). Twelve genomes were incorrectly classified because they were clustered with the outgroups from Nitrosomonadales. As for the phylogeny of the remaining 78 genomes, we proposed that the order should be classified from existing three family-level lineages into five family-level lineages, namely, Rhodocyclaceae, Azonexaceae, Zoogloeaceae, Azospira_f, and Uliginosibacterium_f, with the support of high BS value for each lineage. The classification of the first three families was generally consistent with the phylogenetic analysis on the basis of 16S rRNA gene sequences, whereas the two latter families were not. Containing only two genomes of Azospira oryzae and Oryzomicrobium terrae, Azospira_f that was classified as a member of Rhodocyclaceae based on 16S rRNA gene14 (and Fig. 1) was phylogenomically highly related to Azonexaceae. However, the weakly supported node (BS = 49%) between Azospira_f and Azonexaceae suggested that the former could be designated as a family-level lineage independently. For the proposed Uliginosibacterium_f that contained two genera (Uliginosibacterium and Candidatus Dactylopiibacterium), the rRNA-based phylogeny also supported that the two genera, together with Niveibacterium umoris, formed a clade separated from Zoogloea (Fig. 1). The phylogenomic affiliation of this genus was unclear because of the absence of genomes from Niveibacterium. The only one genome from Azovibrio restrictus (the type species of the genus) presented as a single branch and clustered with the other taxa within Azonexaceae. Thus, we still designated this genome as a member of Azonexaceae in this study. The determination of the taxonomic rank of this clade should require additional related genomes and information. Polyphyletic distribution was observed for type strains in Azoarcus (e.g., DQS4 and ARJX01). Therefore, further clarification should be conducted for the genera.

Maximum-likelihood tree on the basis of 40 concatenated universal genes. Distinctive colors of branches stand for different family-level lineages and outgroups. The colors in the labels stand for sample sources as annotated. The numbers on the side of the nodes show the bootstrap value (n = 500). The right side shows the distribution of five denitrifying genes in Rhodocyclales, with each shape representing a gene. Blank indicates absence and different colors in the shape represent different gene clusters for a given gene.
Strains in Azospira_f and Uliginosibacterium_f were obtained from soil, sediment and host-associated environments and no genomes came from wastewater-related samples. However, Azospira (if correctly classified) has been detected in many AS samples by high-throughput sequencing of the rRNA gene5. The three other lineages, namely, Rhodocyclaceae, Azonexaceae and Zoogloeacea, contained a high proportion of strains from activated sludge. However, the phenomenon should be attributed to the biased contribution of wastewater-related strains in the total genome dataset. The wastewater-related strain as an artificial system must be sourced from other natural environments. Soil and sediment could be the potential sources because the wastewater strains usually have phylogenetically related taxa in these environments (Fig. 2).
Distribution of genes encoding functional traits related to taxonomy
A set of key genes corresponding to phenotypic classification of this order was sought in the genomes. The features included anoxygenic photoheterotrophy, nitrogen fixation, utilization of chlorate, perchlorate and selenate as electron acceptors and reactive oxygen species detoxification54, (Table 1). Species of Rhodocyclus, which is a genus of Rhodocyclales, are anoxygenic photoheterotrophic organisms. Despite the absence of genome from the species R. purpureus, the key genes pufM and pufL were only detected in this genus. The phylogenetic topology in this order (Fig. 2) indicated that this metabolic capability should likely be horizontally gained for this genus (less likely lost in all other clades). Moreover, superoxide dismutase and catalase-peroxidase detected in nearly all genomes suggested the aerobic or aerotolerant property in the majority of this order55. The occasional absence of the two enzymes might be due to the genomic incompleteness. Nitrogen fixation, which was previously recognized in Azoarcus, Azonexus, Azospira and Azovibrio54, was found widely distributed in many groups. For example, all Uliginosibacterium_f strains have complete nitrogen fixation genes in their genomes, whereas this feature has not been experimentally validated in these organisms56.
Respiration on chlorate or perchlorate was previously discovered in strains from Azospira, Dechloromonas and Propionivibrio, whereas selenate could serve as electron acceptor only for T. selenatisa54. Interestingly, the strain of D. agita has been validated in chlorate respiration57, but the functional gene was absent in the complete genome of the non-type strain of the same species, that is, JAET01. Two strains in Zoogloeaceae contained chlorate reductase and eight strains, two from Azonexaceae, one from Azospira_f and five from Zoogloeaceae carried gene-encoding perchlorate reductase. Only A. oryzae and D. hortensis were experimentally verified54. The genome EbN1, which is an invalid taxon Aromatoleum aromaticum previously58, surprisingly contained all three reductases. Therefore, the versatility of this genome on diverse electron acceptors was evident. Our survey based on gene content greatly expanded the strain spectrum that could respire on these electron acceptors. However, a patchy distribution of these genes among families, genera and even within species suggested that they were unsuitable for taxonomic basis.
Genomics revealed distribution and potential horizontal transfer of denitrifying genes in Rhodocyclales
As shown in Fig. 2, narG was detected in all families except for Azospira_f, whereas nirS was completely absent only in Uliginosibacterium_f. However, narG exhibited a patchy pattern, but nirS was widely present in most genomes in the four families. nirK was only found in the three genomes from Ca. Dactylopiibacterium carminicum. nirK and nirS are usually mutually exclusive in most cases, although nir type may differ within closely related taxa21,59. norB and nosZ showed a co-occurred tendency in most lineages, except for many Rhodocyclaceae genomes, which were absent in norB and present in nosZ, as reported by a previous summary on Accumulibacter60. Two strains, namely, A. restrictus DSM23866 and XMAS_Bin103, containing norB but not nosZ, might possibly produce nitrous oxide during denitrifying61. However, the absence of nosZ in the latter might be derived from the low genomic completeness (<75%, Table S1). The previous survey on the co-occurrence of denitrifying genes has shown that strains containing nirS, instead of nirK, are more likely to include all downstream denitrifying genes62. This conclusion could be generally supported by the majority of examined Rhodocyclales genomes except for Rhodocyclaceae. The nirS-containing genomes in this order usually contain the downstream genes of norB and nosZ.
The co-existence of multiple copies of denitrification genes in certain genomes has been observed, especially for nirS63,64. Multiple copies of NIR genes might be related to alternative functions, niche adaption and different selective pressures due to HGT or gene duplication65. Fitness advantages of multiple copies of nirS have been experimentally validated on Thauera spp.66. In the Rhodocyclales genomes, the ORFs of narG, nirS, norB and nosZ could be divided into nine orthologous clusters (designated as NarG1, NarG2, NirS1, NirS2, NirS3, NorB1, NorB2, NosZ1 and NosZ2, Table S3). More than one copy of each gene was detected in many Rhodocyclales genomes (Fig. 2 and Table S3). The multicopy of nirS could be divided into two types. The first was that the copies were within the same gene cluster; thus, they were likely to be generated from gene duplication or gained from closely related taxa. The other type consisted of copies with large divergence (not grouped in a gene cluster). The complex and patchy distribution of multicopy nirS (Fig. 2 and Table S3) suggested frequent gene loss or horizontal gain gene from related taxa during diversifying.
On the one hand, we constructed the phylogenetic tree of NirS covering popular denitrifying bacterial lineages (Fig. S3) to track the potential horizontal transfer events of denitrifying ORFs between Rhodocyclales and other taxa. In the tree of six clades (clades I–VI), nirS sequences from Rhodocyclales dispersedly located in three clades (clades III, V and VI), while they were absent from two root clades (clades I and II). One clade exclusively comprised Gamma- and Alpha-proteobacteria lineages (clade IV). ORFs in the three clades corresponded with the three NirS clusters (NirS1, NirS2 and NirS3). Noticeably, in at least two clades (clades V and VI) that contained taxa from multiple bacterial classes, Rhodocyclales sequences located in the root. Therefore, they or their ancestor taxa might be the donors of horizontal transfer events before the subsequent evolution of the clades. Although a previous study has proposed that nirS is overall congruent with rRNA-based phylogeny with occasional horizontal transfer events65, our results suggested that the events might be frequently detected if additional genomes were presented.
On the other hand, we further profiled potential within-order HGT events for each of the seven denitrifying gene clusters (NirS3 and NorB2 were excluded due to the small number of sequences). As shown in Fig. 3, except for NirS1, all the six other clusters had supporting evidence of interfamily HGT events. Strong evidence of a single HGT event was observed in NarG1, NarG2 and NosZ1, the former two of which were supported by multiple genomes from one family. Thus, their sequences formed a monophyletic branch, which was inserted to a lineage of another family. Multiple events were observed in NirS2, NorB1 and NosZ2, where the true transferring history might be difficult to track. The genomes of DGXX01, DGYB01, AUCE01, AUCH01, LGUD01 and A. oryzae PS exhibited horizontal transfer events in multiple clusters. Given that all these genomes were generated from isolates, the unusual phylogeny might not be derived from genome contamination. As pointed out by Soucy et al.67, HGT between closely related organisms may occur frequently but is difficult to detect owing to the indistinguishable incongruence between the HGT gene and phylogeny. However, the inter-family HGT of the denitrifying gene was detectable and did not occasionally occur within Rhodocyclales. This result indicated that several taxa in the order could gain (or restore) the denitrification capability by acquiring functional genes from the related taxa in the order, which conferred certain superiority in their ecological adaption. A closely related source of HGT gene might have potential merits in functionality and fitness68.
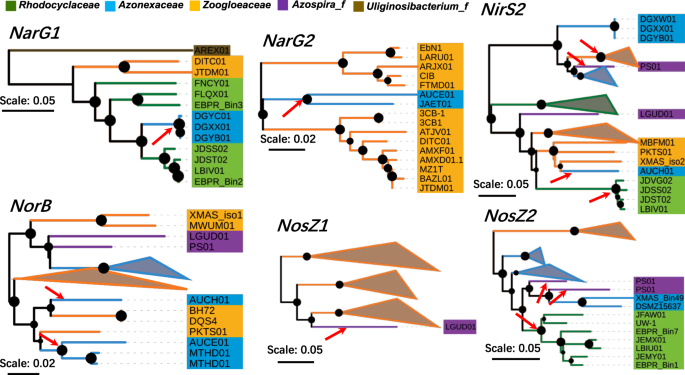
Neighbor-joining trees of denitrifying gene clusters of Rhodocyclales. Six of the seven gene clusters were present with signs of horizontal transfer events. Arrows show the potential horizontal transfer events, where a branch was inserted to a certain lineage of another family. The nodes with BS value over 50% (n = 1000) were present and the size increased with the value.
Distribution of major sub-lineages of Rhodocyclales in global WWTPs were significantly governed by temperature and MLSS
As shown in Fig. 4A, Rhodocyclales accounted for 7.7% of the total bacterial community in WWTPs. This finding was generally consistent with another survey at the global level with few WWTPs5. The three families, namely, Azonexaceae, Rhodocyclaceae and Zoogloeaceae, accounted for 2.9%, 1.1% and 2.6% on average, respectively. Three top OTUs, namely, Accumulibacter sp., Zoogloea sp. and Thauera sp., accounted for 0.3%, 1.1% and 0.4% on average, respectively.

Distribution of Rhodocyclales and the factors governing its distribution in WWTPs. (A) Violin plot showing the abundance distribution of Rhodocyclales in global WWTPs. Only taxa with the median abundance of >0.1% are shown. (B) RDA plot based on Rhodocyclales across 131 WWTPs. The P value shown in the plot was estimated by anova.cca function and each circle represents a WWTP. The red arrows indicate the taxa and the blue arrows indicate the environmental variables with P < 0.05 (envfit function). The top and right panels show the abundance distribution of the Rhodocyclales at the family level along with the first and second constrained axes.
The RDA analysis (Fig. 4B) showed that the abundances of Rhodocyclales, Rhodocylaceae and Accumulibacter OTU were negatively correlated to the latitude and positively correlated to temperature in full-scale WWTPs24. Previous reports on EBPR have shown optimal phosphate removal performance at 10 °C to 20 °C69,70, but these studies have focused on the competition relationship between polyphosphate- and glycogen-accumulating organisms in laboratory EBPR systems with enriched Accumulibacter. Except for one study on 18 WWTPs by Mao et al.71, information on factors governing the distribution of the aforementioned genus in considerably low abundance in full-scale WWTPs is scarce. Our result suggested that the distribution of Accumulibacter could be selected with high temperature under the subdominant situation in full-scale WWTPs, which was similar to that of Mao et al.71.
Zoogloeaceae is negatively correlated with Rhodocylaceae and dominates in low-temperature areas. Currently, no other evidence supports the low temperature adaptive growth advantage of Zoogloeaceae. The production of extracellular polysaccharides (EPSs) in sewage treatment is negatively correlated with temperature72. The advantage of Zoogloea at low temperatures might be related to the increase in EPS production. A possible explanation is that most microorganisms grow relative slowly at low ambient temperature, while the aggregates formed by Zoogloea73 can retain in AS biomass via minimizing the elution process compared with other taxa.
Interestingly, Azonexaceae was positively correlated with the running years of the WWTP and negatively correlated with MLSS. Low MLSS indicated high efficiency in treatment when the operational load and performance were comparable. Therefore, Azonexaceae might be associated with high treating efficiency in wastewater treatment.
Metagenomics suggested predominant role of Rhodocyclales in denitrifying in wastewater treatment systems
Although previous rRNA-based investigations have shown that Rhodocyclales is a top lineage in AS samples5,74, its contribution to the denitrifying gene pool has not been comprehensively characterized. We taxonomically quantified narG, nirS, norB and nosZ in metagenomic data from 44 globally collected AS samples. As shown in Fig. 5, narG was the most abundant in most samples compared with the three other genes because of its long length. SRR1068202, which is an AS sample from a laboratory EBPR bioreactor, showed a very high abundance of nirS and a low abundance of narG. We speculated that the dominant taxa in the system should be Accumulibacter population without narG.
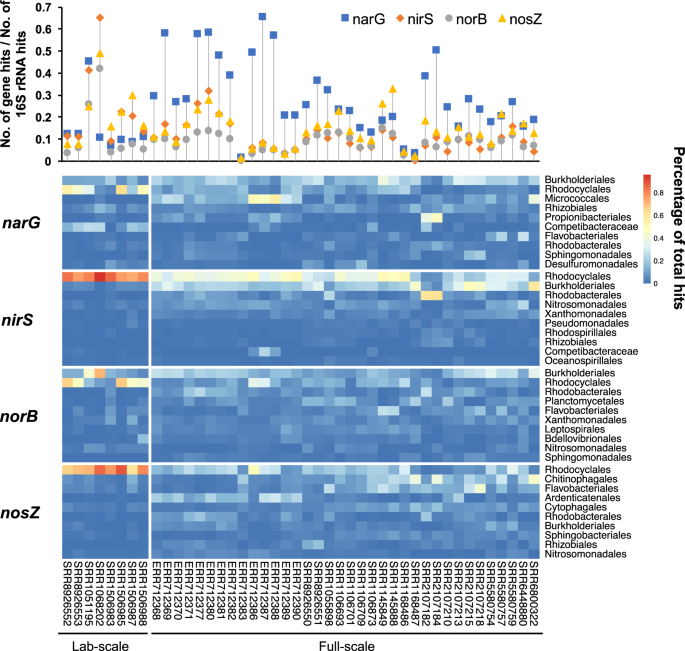
Distribution of four denitrifying genes in 44 activated sludge metagenomes. Dropline diagram on the topside shows the normalized abundance of each 16S rRNA gene. For each gene, the relative abundances of the top 10 orders (sorted by the mean value of relative abundance for each gene) are displayed in the heatmap.
Rhodocyclales-derived denitrifying genes were all ranked at the top or the second among all orders. Therefore, they significantly contributed to the denitrifying process. Eight laboratory reactors were likely dominated by this order based on the datasets possibly due to a biased sampling from EBPR systems. For 36 full-scale samples, Rhodocyclales-derived nirS and nosZ were ranked as the top in 33 and 21 samples, respectively, whereas the numbers were 9 and 10 for narG and norB, respectively. Another order in Betaproteobacteria, that is, Burkholderiales, was also an important contributor to potential denitrifiers because it ranked as the top for narG and norB, followed by nirS. Its abundance was close to that of Rhodocyclales according to a 16S rRNA survey on multiple globally collected AS samples5,74. However, this order only slightly contributed to nosZ (ranked at the seventh order).
Metagenomic survey on a few AS samples has confirmed the contribution of Rhodocyclales in the denitrifying community19. Surprisingly, no comprehensive PCR-based survey on the diversity of denitrifiers in AS has been performed. A recent study performed by Ma et al.75 updated the primers for detecting environmental denitrifiers, which would facilitate the study of denitrifier diversity in AS samples together with the metagenomic approach, as applied by the present study. Further combination of transcriptional and biochemical studies on the denitrifying process in full-scale AS is important to understand the true functional contribution and operational parameters related to their functions.
Source: Ecology - nature.com
