Microbial community profile based on marker genes
We de novo assembled 27 metagenome samples, the reads from which were previously used in a study that involved mapping to 505 genomes reconstructed from prior data sets to link organisms to groundwater of different depths [12]. In the current study, we extracted assembled sequences of rpS3 and used read mapping to scaffolds carrying this gene to follow organisms over the 5-day eruption cycle. This approach allowed us to track 914 putatively distinct microbial species (Fig. 1), greatly exceeding the 505 previously reconstructed genomes [12].
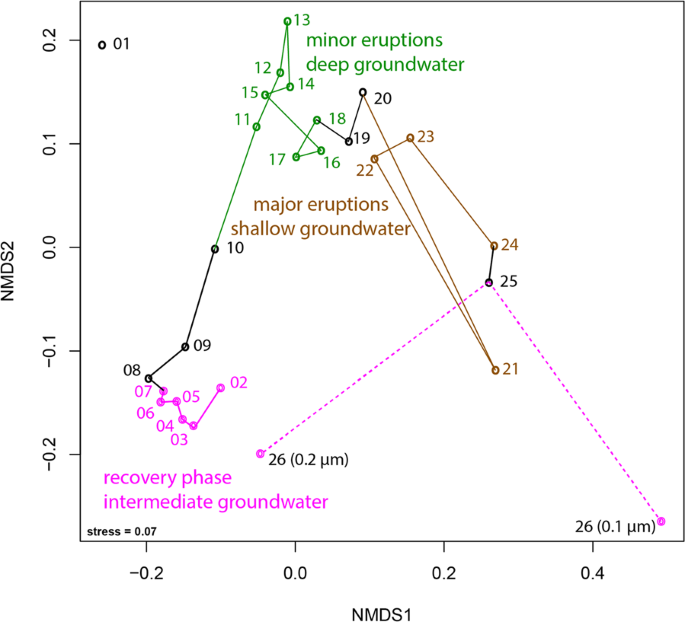
Nonmetric multidimensional scaling based on the Bray–Curtis index. The connections show the trajectory of the different samples taken throughout the eruption cycle. Sample 01 was not included as it was an amplified library due to low biomass (see “Material and methods” for further details). Sample 26 was collected after the end of the major eruptions and is already part of the recovery phase (thus colored in pink). Black color indicates samples that were collected during transition between phases. Please note, that the sample was also size-fractioned into a 0.2-µm and a 0.1-µm filter. For details on individual rpS3 abundances please see Fig. S5 and Table S5.
We detected a large community shift associated with different eruption phases. According to previously published geochemical data [12], the first phase, referred to as the recovery phase, sources groundwater from an aquifer of intermediate depth, likely a Navajo Sandstone-hosted aquifer. During the second minor eruption phase, water from a deeper aquifer is sourced (likely Wingate Sandstone-hosted) and during the third major eruption phase, an increased fraction of shallow groundwater is sourced (Fig. S1). Grouping of samples into different clusters in an ordination analysis based on community composition (Fig. 1) revealed stepwise changes throughout the eruption cycle. The final sample, which was taken after the end of the major eruption phase and as the geyser transitions into the next recovery phase was size-fractionated, with cells collected sequentially on a 0.2 µm filter and followed by a 0.1 µm filter (sample 26, Fig. 1). The community composition on the 0.2 µm filter plots near samples from the beginning of the first cycle in the ordination analysis, indicative of a restoration of the initial microbial community (Fig. 1).
In situ carbon fixation sustains microbial communities irrespective of aquifer depth
Previous community-wide genomic analyses suggested that carbon fixation might sustain the relatively complex aquifer microbial communities, but direct evidence was lacking [10, 33]. We measured the stable carbon isotope composition (i.e., δ13C values) of IPL-derived bacterial FA and archaeal phytane. The values for 14 samples were plotted as a function of sampling time and compared with the δ13C values of DIC and CO2 in the ecosystem (Fig. 2a, b). The δ13C values for DIC sampled from the geyser discharge over its 5-day cycle ranged from 3.6 to 8.0‰ (average = 5.0‰, std. dev. = 1.4‰) and showed no systematic variation with relative depth of source water (Fig. S6). The δ13C values for phytane range between −47.0 and −32.8‰ and for bacterial lipids (expressed as weighted average of all FAs) from −32.7 to −22.1‰. We found very little genomic evidence for utilization of methane [35] by these communities and methane was not detected in the geyser gas emissions [12]. Thus, we do not attribute the 13C-depletion of phytane to methane metabolism by methanogens/methanotrophs. Alternatively, heterotrophy could sustain microbial metabolism in the aquifers sourcing Crystal Geyser. However, the Wingate and Navajo aeolian sandstone aquifers have little associated sedimentary organic carbon [40, 41] that could serve as substrate. Similarly, dissolved organic carbon (DOC) concentrations in minor eruption phase fluids (~1 ppm, Table S3) are overall similar to global median groundwater [42], suggesting no significant admixture of exogenous DOC, for example from nearby oil reservoirs. It is possible that advection of exogenous DOC is more prevalent during major eruptions, but no DOC samples could be obtained from this phase. Still, the 13C-depletion in FA and phytane suggests that the majority of biomass is not primarily derived from heterotrophic incorporation of DOC during minor eruptions: phytane (δ13C is −42 to −47‰) and the C16:0, C16:1w7, and C18:1w7 FA are too depleted in 13C (δ13C is −29 to −34‰) to originate primarily from DOC (δ13C is −19 to −24‰, Table S3), while only the δ13C value of C18:0 FA (−27‰) is compatible with the small fractionation between substrate and FA observed in heterotrophic bacteria [43]. Importantly, the DOC in Crystal Geyser aquifers could be derived from in situ primary production and thus sustain heterotrophic bacteria.
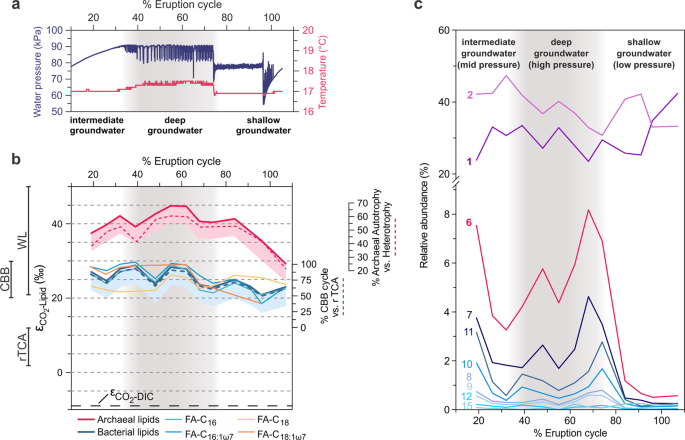
a Water pressure and temperature over the geyser cycle showing sourcing of fluids from the conduit (mixed), the deep aquifer, and the shallow aquifer from Ref. [12]. b Stable carbon isotope fractionation of archaeal lipids (phytane, released from archaeol), individual bacterial fatty acids (FA, released from diacylglycerols), bacterial lipids (weighted average of FA), and dissolved inorganic carbon (DIC) relative to CO2 (εCO2-Lipid) over the geyser cycle. Lines to the left of the panel show expected ranges of εCO2-Lipid (accounting for up to 5‰ additional 13C-depletion of lipids relative to biomass, indicated by shaded areas) for the Calvin–Bassham–Benson (CBB; [46,47,48]), the reductive tricarboxylic acid cycle (rTCA [46, 49, 50]), and the Wood–Ljungdahl pathway (WL, reductive acetyl-coenzyme A pathway; [39, 62, 63]). The blue dashed line indicates relative contribution of carbon fixation through the CBB cycle versus the rTCA cycle for bacterial lipids (assuming maximum fractionation due to high in situ [CO2] and [DIC]). The red dashed line indicates the relative contribution of autotrophy versus heterotrophy (uptake of bacterial CBB/rTCA-fixed carbon) to archaeal lipid biomass, calculated from mass balance of δ13C values of bacterial and archaeal lipids (assuming maximum fractionation for archaeal autotrophy due to high in situ [CO2] and [DIC]). c Relative abundance of unsaturated diacylglycerol membrane lipids (the number indicates the sum of double bonds in both acyl chains). The distribution is dominated by mono- and di-unsaturated diacylglycerols but polyunsaturated lipids (6–15 unsaturations) increase markedly in deep aquifer fluids. Grey shading indicates major eruptions, which source deep aquifer water under high pressure.
Stable carbon isotopic compositions of lipids point to a predominantly autotrophic origin of microbial biomass. Due to the high in situ concentration of both HCO3− (69–84 mmol/L; [44]) and CO2 (at saturation level throughout the geyser [12, 44]), maximum fractionation by carbon-fixing microorganisms in the geyser can be assumed [45]. Changes in inorganic carbon speciation and thus fractionation are unlikely, as HCO3− concentrations, temperature (~16.8–17.5 °C, Fig. 2a), ionic strength (15–19 mS/cm, Fig. S1), and pH (6.4–6.5, [44]) stay in narrow ranges. In addition, growth rates of Crystal Geyser communities are likely to be low and thus carbon isotope fractionation (ε) would be expected to be maximally expressed. Based on this, and the known range of ε for carbon fixed via different pathways [46,47,48,49,50,51], it is plausible that the majority of archaeal lipids were synthesized via the Wood–Ljungdahl (WL, reductive acetyl-CoA, εDIC-lipid > 30‰) pathway from DIC, with εDIC-lipid of 38.3–53.9‰ observed in phytane derived from archaeol-based IPLs (Fig. 2b). This is in accordance with previous investigations of Crystal Geyser, which reported dominance of Altiarchaeota in the deepest aquifer [12]. Altiarchaeota fix carbon via a variant of the WL pathway with a fractionation εDIC-lipid of ~63‰ [52] (assuming εDIC-CO2 as ~10‰ at 15 °C calculated after Ref. [53] and δ13C of archaeal lipids from a 99% enrichment of Altiarchaeota reported in Ref. [52]). The observed εDIC-lipid values for archaeal lipids in many samples are below the maximum theoretical fractionation, implying that archaea in Crystal Geyser are not exclusively autotrophic but also take up isotopically heavier organic carbon. One likely source is archaeal utilization of organic carbon fixed by bacteria via the Calvin–Benson–Bassham (CBB) and reductive tricarboxylic acid cycle (rTCA) cycles, which would be more enriched in 13C than carbon fixed via the WL pathway. The degree of heterotrophic uptake by archaea can be approximated using a mass balance calculation involving mixtures of carbon with (i) the maximum theoretical fractionation for autotrophic archaeal carbon fixation via the WL pathway and (ii) the observed δ13C values of bacterial lipids (accounting for up to 5‰ additional 13C-depletion of lipids relative to biomass). This calculation would imply that archaea are predominantly autotrophic in deep groundwater (up to 70% of the biomass carbon fixed through WL pathway), but in the intermediate and shallow groundwater form up to 69% of their biomass by taking up bacterial organic carbon fixed through the CBB and rTCA cycles (Fig. 2b).
Bacterial lipids display the carbon isotopic fractionation expected from the CBB cycle relative to CO2 (εCO2-lipid of 20.9–28.8‰ observed vs. 30‰ theoretical) and not that expected from fixation via the rTCA cycle (εCO2-lipid < 12‰ theoretical). Sequences encoding the CBB pathway are fairly abundant in the ecosystem throughout the recovery phase [12] and likely contributed to the bacterial lipid pool of samples collected during that period. This agrees with previous genomic findings that identified several highly active iron-oxidizing Gallionella species carrying this pathway [12]. Importance of Gallionella in Colorado Plateau aquifers is further indicated by the association of organic carbon with fossilized Gallionella cells in postdepositional iron concretions of the Navajo sandstone [40]. However, genomic analyses suggested that one of the most abundant organisms in the shallow aquifer (Sulfurimonas sp.) fixes carbon via the rTCA cycle [12]. From mass balance calculations using the observed and theoretical fractionations, we estimate that carbon fixed via the rTCA cycle contributes as little as 12% to the bacterial biomass in the deep and intermediate aquifer but up to 78% of the biomass in the shallow aquifer (Fig. 2b). Overall, the observed carbon isotopic composition of the bacterial lipids could be explained as the result of a mixture of Sulfurimonas-derived lipids and lipids formed via the CBB pathway.
Degree of unsaturation of bacterial IPL changes with groundwater source depth
Using in-depth analyses of IPLs we tracked the abundance of IPL-bound bacterial unsaturated FAs across the eruption cycle. The unsaturations presumably correspond to double bonds but due to the mode of detection, we cannot strictly rule out cycloalkyl groups found in FAs of some bacteria [54], although typically not in higher numbers than one per FA. Interestingly, the relative abundance of highly unsaturated FAs correlated with the groundwater depth source (Fig. 2c). The cumulative abundances of IPLs with one or two double bond equivalents in their FA side chains were fairly consistent throughout the cycle, indicating little variation between the different groundwater sources. However, IPLs with seven or more unsaturations, i.e., at this high number presumably double bonds, were relatively abundant during the first phase, when groundwater was sourced from intermediate depths. These lipids were even more abundant during the middle phase, during which groundwater derives from the greatest depth, and almost undetectable in samples collected in the final shallow groundwater eruption phase. One explanation for elevated abundance of polyunsaturated lipids is their derivation from eukaryotes [55]. The occurence of tentatively identified DGCC-type (1,2-Diacylglyceryl-3-O-carboxyhydroxymethylcholine) betaine lipids is unprecedented in bacteria and supports the presence of Eukaryotes in the ecosystem, although the pathway for generating these lipids and its phylogenetic distribution remains unknown [56]. In general, Eukaryotes have been found in the geyser [57], primarily in a sample of decayed wood added to the geyser conduit, and they have been detected by rpS3 analysis in the current study. However, they are not very abundant, and fluctuate heavily throughout the cycle (Fig. S7). Due to the pronounced abundance maxima during deep aquifer eruptions, the most likely explanation for the presence of polyunsaturated FAs is their origin from organisms adapted to high pressures in the deeper subsurface. Bacteria are an additional, potential source of polyunsaturated FA, as the biosynthetic capacity for these lipids is widespread in terrestrial and aquatic bacteria such as Shewanella, Vibrio, and Geobacter spp. [58,59,60]. Incorporation of double bonds in bacterial FAs is a well-known mechanism that increases membrane fluidity at high pressure and low temperature [61, 62]. Consequently, a great diversity of unsaturated FA biosynthesis gene sequences are found in the Crystal Geyser metagenomes. For instance, we detected 1959 different protein clusters (>10% dissimilarity) of 3-oxoacyl reductases, representing 11,548 protein sequences in total (Fig. S8). As temperature remained nearly constant at around 17 °C (Fig. 2), high-FA unsaturation could represent an adaptation to the high pressures faced by indigenous bacterial communities in the intermediate and deep aquifers, supporting a direct link between groundwater sources and lipid profiles.
Predicting linkage of IPLs to uncultivated organisms
We detected 295 different IPLs in the 14 lipidomes but a strict organism-lipid relation was unresolved due to the complexity of the community. Assignment of lipids to specific organisms is further complicated by the existence of multiple potential source organisms for common lipid types and the distinct characteristics of a low-energy habitats in the subterranean aquifers. Distinct turnover times of lipids and DNA as well as lipid recycling, which may be a common strategy utilized by energy-starved archaea in the subsurface [63,64,65], could adversely affect correlations. While relative turnover times of DNA and lipids remain unconstrained, the predominance of chemically labile phosphoester IPLs in Crystal Geyser facilitates comparatively faster turnover of lipids compared with marine deep biosphere environments where ether-based IPLs, including glycolipids, are prevalent [66, 67]. Irrespective of whether they represent snapshots of a dynamic system or signals accumulated over longer timescales, the systematic changes in metagenomes and lipidomes indicate distinct, stratified habitats within Crystal Geyser.
In the current study, we used a time series of 14 metagenomic and coupled lipidomic data sets to establish correlations between marker gene abundances and IPLs. Based on this analysis, we tested for evidence for the assignment of lipids to organisms. Specifically, relative abundance patterns of individual organisms were correlated with the relative abundance of the 295 IPLs (only organisms and lipids were considered if they were identified in at least seven out of fourteen samples). Lipids were also co-correlated with other lipids and primary and secondary lipid assignments were investigated via a network analysis (Fig. 3). Although the majority of IPLs were found to be unspecific, significant correlations were observed between a subset of lipids and organisms: 44 primary lipids correlate significantly with 22 different marker genes (organisms) and 63 secondary lipids (Table 1).
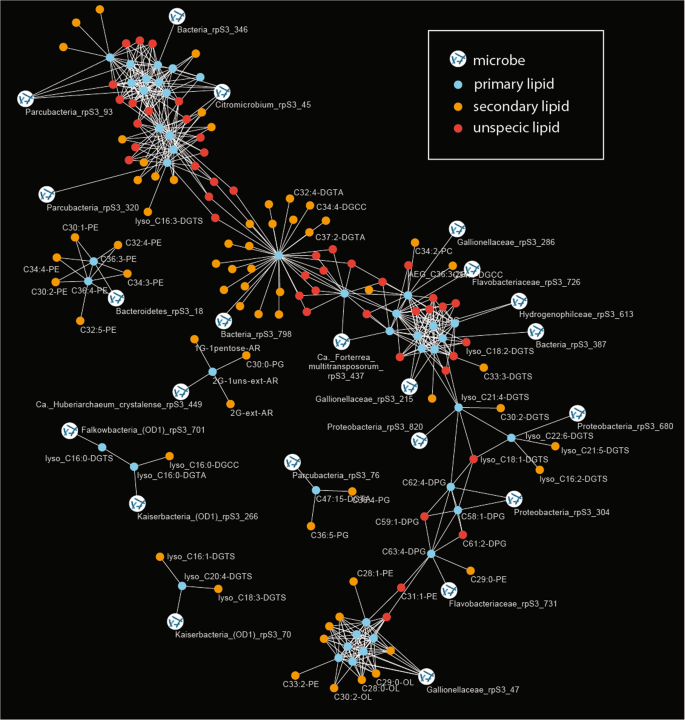
The primary lipids were defined based on a direct correlation of their relative abundance with rpS3 gene abundance (Bonferroni-corrected p value < 0.005). Secondary lipids showed a significant correlation with primary lipids and are indicative of a biological connection between the lipids (e.g., lipids from microbial symbionts or co-correlated organisms). Unspecific lipids shared primary lipids with different organism assignment. Due to visual limitations only few IPL names are displayed in the figure; all organisms to lipid correlations are provided in Table 1, raw data can be accessed in Tables S4 and S5.
It is important to note that all significantly correlating ether-based isoprenoid lipids were assigned to archaea (Ca. Huberiarchaeum crystalense) as this provides confidence in the correlation-based approach. However, it is unclear whether correlation of the main lipid of the Ca. H. crystalense and one bacterial lipid is a spurious covariation or if this represents assimilation of a bacterial membrane lipid by archaea (Huberiarchaeum did not correlate with that bacterial lipid; Table 1). Of particular interest were the lipids of Altiarchaeota, since these had been characterized earlier [52]. These previously detected lipids, including hexose-pentose archaeol (1G-1pentose-AR; for mass spectrometric identification see Fig. S4) and dihexose extended archaeol (2G-ext-AR), were the most abundant archaeal lipids in the current study but most abundances showed little correlation with the Altiarchaeota abundances. On the one hand, this might be due to the presence of multiple different strains of Altiarchaeum sp. in the samples (based on rpS3 genes; Fig. S9), which can harbor different lipid profiles as shown previously [68]. On the other hand, the main archaeal IPL (2G-AR) was also present in the sample filtered through a 0.2-µm filter and collected onto a 0.1-µm filter but Altiarchaeum sp. DNA was not (based on rpS3 genes). This indicates the lysis of Altiarchaeum sp. during the filtration process, possibly due to oxygen stress, a resistance that Altiarchaeota in Crystal Geyser apparently do not possess [52]. Altiarchaeota in Crystal Geyser also have Ca. H. crystalense as a symbiotic partner [69], which could derive its lipids from the Altiarchaeota and was indicated to possess genes, whose products might be involved in lysis of Altiarchaeota cells [12]. In addition, longer turnover times of the chemically stable ether-bound lipids of archaea [66, 67] compared with DNA could deteriorate correlations. Nevertheless, some IPL signatures (e.g., 2G-ext-AR) showed a significant correlation with the sum of rpS3 abundances of all Altiarchaeum sp. in the sample, supporting the above-mentioned assumptions (Fig. S9).
We detected one low abundance archaeal lipid, an unsaturated variant of 2G-ext-AR (2G-1uns-ext-AR), which had not been identified in Altiarchaeota. This may be a previously unrecognized membrane component of Altiarchaeota or derived from another archaeon. Its abundance correlated only weakly with other Altiarchaeota lipids but highly significantly with the abundance of Huberiarchaeum, thus it may derive from this organism. Huberarchaeota are the second most abundant archaea after Altiarchaeota in this ecosystem and they are predicted to have the genes required to synthesize lipids from scavenged isopentenylpyrophosphate [12]. The molecular structure of 2G-1uns-ext-AR differs by only one double bond from the Altiarchaeota lipid 2G-ext-AR, so Huberarchaeota may largely derive its lipids from Altiarchaeota, which was suggested to be its host [12]. The relative abundance of 2G-1uns-ext-AR correlated significantly with 2G-ext-AR, highlighting the potential biological meaning that can be inferred from IPLs, whose abundances do not correlate with certain organisms but with certain lipids instead. Given the confident assignment of 2G-1uns-ext-AR to Huberarchaeota, we used the p value for that assignment as a conservative correlation p value for further predictions (Bonferroni-corrected p value < 0.005), which are presented in Table 1.
Several bacterial groups were correlated with the occurrence of cardiolipins (diphosphatidylglycerol (DPG) lipids), which are involved in osmotic stress response, membrane ordering, and regulation of cell curvature [70,71,72,73]. Specifically, DPGs are required for maintaining cell shape in rod-shaped bacteria [71]. Consequently, DPGs found in Crystal Geyser are correlated with clades typically forming rods or elongated cell shapes, including the Flavobacteriaceae and Gallionellaceae (Table 1). These matching correlations thus further validate our statistical approach.
Lysolipids and Candidate Phyla Radiation bacteria
In order to investigate lipids of bacteria from the CPR [13], we analyzed the IPLs of a small cell size fraction collected on a 0.1-µm pore-size filter after 0.2-µm pre-filtration. Based on the corresponding metagenome, the sample contained 186 different organisms, 165 of which were classified as CPR based on rpS3 sequences and one low abundant organism was classified as a member of the DPANN radiation (Ca. H. crystalense). Surprisingly, the most abundant organism in the sample based on metagenomics was a Sulfurimonas, which apparently passed through the 0.2-µm filter (read mapping-based coverage in 0.2-µm filter was 8.4 in the corresponding 0.1 µm filter 1081.9). We identified 72 different IPLs in the post-0.2-µm sample, all of which were acylglycerols. Consequently, the CPR organisms in this sample must possess FA-based lipids. This is important because the composition of lipids of CPR bacteria is unknown. Interestingly, 22 of the 72 lipids (31%) were lysolipids, all of which contained betaine headgroups (for structural characterization see Fig. S3). By contrast, these lipids constituted only 18% across the entire sample set. Cultured bacteria only contain a small fraction of lysolipids, e.g., Sulfurimonas has been reported to only contain a single lysolipid with ~4% abundance [74]. Further, the abundances of several CPR bacteria also correlated significantly with the abundance of specific lysolipids (Table 1).
To further investigate the lysolipid content of CPR bacteria, we selected a sample taken during the recovery phase of the geyser, when little amounts of Sulfurimonas are present as indicated by metagenome sequencing [12]. For this sample cells that passed through a 0.2-µm filter were collected onto a 0.1-µm filter for subsequent metagenomic sequencing and infrared spectromicroscopy. Metagenomic sequencing analysis of the selected sample (CG10_big_fil_rev_8_21_14_0.10; [12]) showed a high abundance of CPR (rank abundance curve in Fig. S10) occupying the first seven ranks of the community. To test for the abundance of lysolipids in this CPR-rich sample, we performed FTIR analysis of the cells (Fig. 4a) and compared the results against a set of reference spectra (Fig. S11). For the first PCA in the 3050–2800 cm−1 spectral region dominated by the aliphatic chains of the lipids, ~85% of the spectral variance is explained by the first five loading vectors (Fig. 4b). Here, the first loading vector contains 55% of the variance, with features that are similar to palmitic acid; with the asymmetric stretching of the CH2 peak centered at 2916 cm−1 (Fig. 4). The peak corresponding to the CH3 asymmetric stretching vibration was used to evaluate the nature of the polar head. The position at 2951 cm−1 of the PC1 is in accordance with the one of lyso-phosphatidylcholine, whereas the peak for phosphatidylcholine is sharper and centered at 2957 cm−1. The corresponding heatmap of the PC1 scores (Fig. 4c) shows the presence of hotspots, a few microns in diameter. The remaining 2, 3, and 4 loading vectors, which explain 18, 10, and 2% of the variance, respectively, show different CH3 to CH2 ratios, and PC3 in particular can be assigned to free FA. In contrast, although the fifth loading vector accounts for only 1% of the variance, its spectral features can be assigned to highly branched and unsaturated lipids similar to those of archaea (Fig. 4b, c; Refs. [14, 75]; see Supplementary material for additional results). This agrees with the presence of DPANN archaea as the second most prominent group of organisms in this sample based on metagenomic profiling (Fig. S10). The combination of the detailed analysis of the IPLs and infrared imaging of two independently sampled small cell fractions suggest that a substantial fraction of some CPR cell membranes consists of lysolipids.
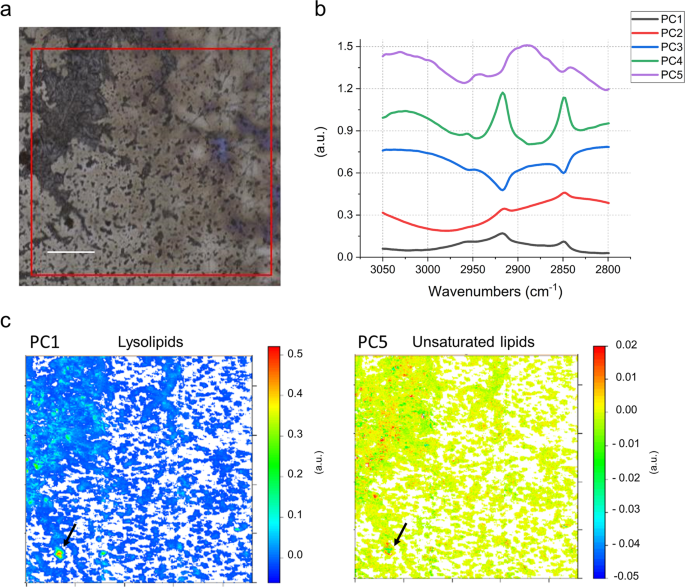
a Field of view in FTIR, 1 × 1 mm (red square). b First five PCA loadings accounting for ~90% of the variance. They describe the directions of maximum variability of the analyzed system. The figure presets the first five vectors, that spectroscopically can be assigned, by similarity of shape and band position, to different types of lipids. c False color maps representing PCA scores PC1 and PC5, respectively. These maps show how the different lipids represented by the eigenvectors in (b), are distributed in the sample. The comparison of the spectral features of the loadings and the reference spectra in Fig. S11 allow assignment of PC1 to lysolipids and PC5 to unsaturated/branched lipids. The arrows point to a hotspot of cells indicating a particularly high distribution of lysolipids (PC1), surrounded by several smaller hotspots of unsaturated/branched lipids (PC5). Given the micrometric lateral resolution of the image (each pixel is 2.6 µm) it is possible to hypothesize that there is a small group of cells in the hotspot area, which is characterized by distinct membrane lipid composition. This can also be observed in other spots throughout the measured biomass. Loadings of the PCA over the whole 900–3700 cm−1 spectral range are provided in Fig. S12. Scale bar 200 µm.
Genome-resolved metagenomics generated 206 new genomes from the entire sample set. Together with 1215 previous genomes [12, 35], our data set included 675 genomes of CPR bacteria that were used to comprehensively investigate their potential for lipid biosynthesis (accessible through https://ggkbase.berkeley.edu/genome_summaries/1491-Bacterial_membrane_lipids_AJP). We found that the CPR genomes do not encode for any known, complete bacterial lipid biosynthesis pathway, yet CPR bacteria are known to have a cytoplasmic membrane based on cryogenic-transmission electron microscopy studies [14]. Interestingly, some members of the Nealsonbacteria phylum (Parcubacteria superphylum) have near-complete pathways for FAs and phospholipid synthesis. They possess some homologs of the FA synthase type II (FAS-II), the main FA biosynthesis pathway in most bacteria. However, they lack the FAS-related acyl carrier protein (ACP) processing machinery (ACP synthase and malonyl-CoA:ACP transacylase). ACP is a peptide cofactor that functions as a shuttle that covalently binds all FA intermediates. Although they lack key genes for FA synthesis, we cannot rule out this group could potentially synthetize FAs by an ACP-independent pathway, as suggested for some archaea [76]. We also searched theses genomes for genes coding for glycerol-3-phosphate (G3P) dehydrogenase, an enzyme responsible for the stereochemistry of the glycerol units of their membrane lipids, and acyl-ACP transferases responsible for the formation of ester bonds between FAs and G3P backbone in phospholipid synthesis. There are two families of acyltransferases responsible for the acylation of the C1-position of the G3P. The PlsB acyltransferase primarily uses ACP end products of FA biosynthesis (acyl-ACP) as acyl donors. The second family involves the PlsY acyltransferase and is more widely distributed in Bacteria. PlsY uses as donor acyl-phosphate produced from acyl-ACP by PlsX (an acyl-ACP:PO4 transacylase enzyme). The acylation in the C2-position of the G3P is carried out by the 1-acylglycerol-3-phosphate O-acyltransferase (PlsC). Screening the Nealsonbacteria genomes, we did not detect any homologs of the first family of acyltransferase, PlsB. However, we identified PlsY and PlsC, but not PlsX. Absence of PlsX raises the question of the enzyme or mechanism for production of acyl-phosphate needed to activate PlsY. Overall, mechanisms or enzymes that produce and/or require ACP were not identified in CPR genomes in this study. Even though this finding opens the possibility for the presence of ACP-independent pathways for FA and/or lipid synthesis in these CPR bacteria, we cannot conclude with confidence that few of these organisms can synthesize lipids de novo. Thus, we suggest that most CPR bacteria derive their membrane lipids, including lysolipids, from coexisting bacteria. Given the small cell size of CPR, lysolipids may be preferred due to their role in reducing membrane curvature stress (e.g., Ref. [77]). As lysolipids can form during lipid breakdown (e.g., mediated by phospholipase A [78]) and can be taken up by other bacteria [79], their utilization by CPR may indicate uptake from degraded bacterial biomass or direct derivation from host cells.
Model of lipid transfer in the community and conclusions
Our approach combined detailed metagenomics with whole community lipidomics and infrared spectroscopy and was informed by isotopic measurements that were constrained by detailed understanding of the geological context. The objective was to probe the carbon cycle within the subsurface microbial ecosystem, particularly the source of fixed organic carbon, but also to investigate evidence for its redistribution into other organisms, especially putative symbionts. Although sample limitation resulted in a lower resolution of isotopic analyses compared with metagenomics, carbon isotope systematics of archaeal and bacterial lipids confidently support the metagenomic predictions that microbial biomass is mostly of autotrohpic origin in all aquifers sampled. Particularly, our results provide evidence that predicted autotrophs were fixing CO2 in situ, using the WL (Altiarchaeum), rTCA (Sulfurimonas), and CBB cycles (Gallionella).
Using lipidomics and infrared spectroscopy on size-fractionated cells, we demonstrate that CPR bacteria with small cell size possess FA-based IPLs, although the corresponding genomes do not encode for a known pathway to synthesize them. Similarly, Huberarchaeota, potential symbionts of Altiarchaeota, were predicted to possess altered archaeal lipids related to those of their putative hosts. Our results support the notion that organisms of the CPR and DPANN radiation do not only scavenge (or symbiotically receive) molecular building blocks or even intact lipids from other bacteria and archaea but also use the corresponding lipids and introduce modifications (Fig. 5).
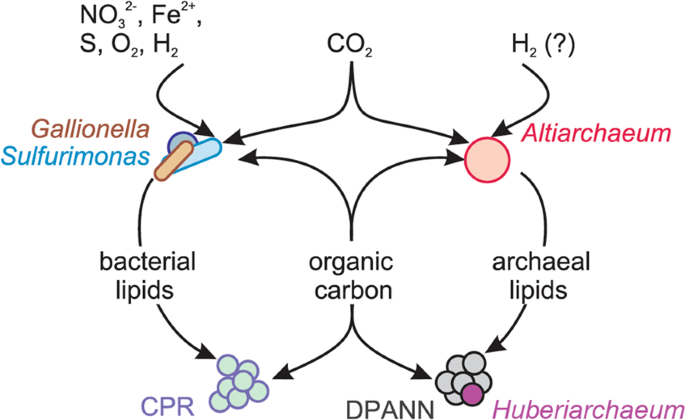
Organic carbon and lipids are produced by Gallionella, Sulfurimonas, Altiarchaeum spp. or other autotrophs, redistributed through the ecosystem and acquired by other community members including CPR bacteria and DPANN archaea.
Source: Ecology - nature.com
