Study area
The Hunter River estuary (151.8°E, 32.9°S) is situated on the temperate southeast coastline of NSW, Australia (Fig. 1). The Hunter River estuary is a typical wave-dominated, mature barrier estuary23 with a large tidal pool that extends 60 km inland to its tidal limits. The Hunter River estuary has semi-diurnal tides and a mean tidal range of approximately 1.2 m. Catchment inflows to the estuary via the Hunter River and its two main tributaries – the Paterson and Williams Rivers – are regulated by dams and weirs.
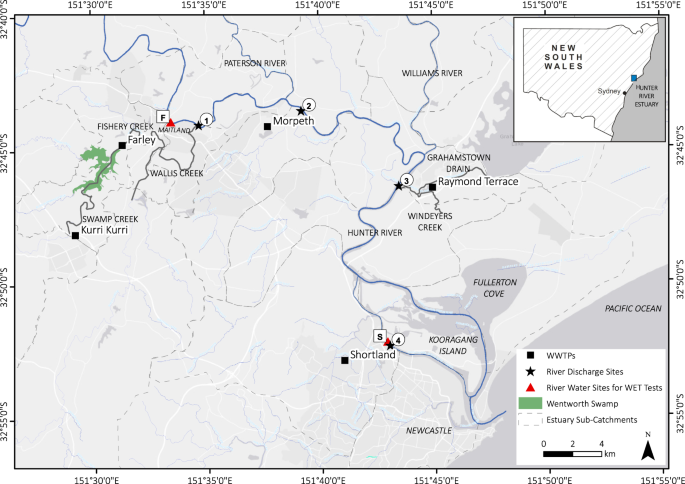
Hunter River estuary study area and WWTP river discharge sites for Kurri Kurri, Farley, Morpeth, Raymond Terrace and Shortland shown as circled numbers. River water collection sites used for WET test dilutions are shown as boxed letters.
The Hunter River catchment covers an area over 22,000 km2 and is typical of many other developed coastal regions globally in that it has been extensively modified by human activity and multiple land uses24. The upper catchment is predominately agricultural land, whereas the lower catchment around the Port of Newcastle includes extensive urban and industrial areas, entrance dredging and training, the world’s largest coal export terminal and a growing multi-purpose cargo hub. Despite these pressures, the Hunter River estuary still supports significant areas of estuarine habitat such as mangroves, saltmarsh, inter-tidal and sub-tidal soft sediment shoals, as well as a Ramsar listed wetland of international significance25.
The Hunter River estuary receives diffuse water pollution and nutrients from the catchment, as well as high nutrient point loads from several major wastewater treatment plants (WWTP) capable of servicing a population of approximately 200,000 people across the floodplain. Generally, these WWTP provide tertiary-level wastewater treatment designed to remove excess nutrients of nitrogen and phosphorus. Unallocated treated effluent is discharged either directly, or indirectly via tributary channels, to the tidal zone of the Hunter River. The tributary channels in this study are freshwater creek systems as they are excluded from tidal flows via one-way floodgates and form part of the expansive Lower Hunter Flood Mitigation Scheme.
The study sampling points were located in the upper, mid and lower portions of the Hunter River estuary (Fig. 1). In the upper Hunter River estuary, the Swamp-Fishery-Wallis Creek system near Maitland, receives unallocated treated effluent from the townships of Kurri Kurri (3.4 ML/day) and Farley (5.6 ML/day). Kurri Kurri WWTP discharges into Swamp Creek which flows into Wentworth Swamp, a large, low-lying permanent waterbody, which in turn discharges into Fishery Creek. Farley WWTP discharges into Fishery Creek downstream of its confluence with Wentworth Swamp, and this flows into Wallis Creek which discharges into the Hunter River estuary (Fig. 1, river discharge site 1). Unallocated treated effluent from Morpeth WWTP (10 ML/day) is discharged directly into the Hunter River, approximately 3 km downstream of Wallis Creek (Fig. 1, river discharge site 2). Unallocated treated effluent from the Raymond Terrace WWTP (7.3 ML/day) is discharged to the Hunter River via Grahamstown Drain and Windeyers Creek (Fig. 1, river discharge site 3). The Shortland WWTP (9.6 ML/day) discharges unallocated treated effluent directly to the Hunter River South Arm (Fig. 1, river discharge site 4). A description of site characteristics for each WWTP river discharge site (Fig. 1) is provided in Table 1.
Microbial field surveys
Water samples were taken on two (2) separate occasions in the Austral summer (November 2016 and February 2017) from five (5) WWTP outfalls along the Hunter River estuary, including Kurri Kurri, Farley, Morpeth, Raymond Terrace and Shortland. Water samples were collected upstream and downstream of each outfall at different distances totalling 20 sampling points (see Table 2). Note that water at Shortland was only sampled from the river immediately upstream of the Shortland WWTP outfall – downstream sampling was not logistically possible and not relevant as the WWTP was not discharging at the time of the investigation. All 20 sites were sampled within the same two-day time window on both sampling occasions with three (3) replicates collected per site for sequencing of microbial (prokaryotic and eukaryotic) communities and a single replicate for water quality measurements.
Specifically, surface water was collected in 2 L sterile Whirl-Paks® stored on ice in the dark until filtering within 24 hours. To capture all microbial cells and fragments in the water samples for DNA extraction and sequencing, samples were homogenised by repeated inversion and 500 mL of water was filtered through a 0.22 µm Express PLUS Polyethersulfone membrane (Millipore) using a hydraulic pump. Filter units were sterilised before use and rinsed with ethanol between water samples. In some cases, when the water samples contained excess organic material, the filters were clogged before 500 mL could be filtered, and the volume that had been filtered was noted for later adjustment of the data. Filters were rolled, inserted into bead tubes from the DNeasy PowerWater Kit (Qiagen) and frozen at −80 °C until DNA extraction and sequencing.
For water quality measurements, water samples were simultaneously obtained using standard bottles provided by a NATA accredited facility for analysis of total suspended solids (TSS), total organic carbon (TOC), total nitrogen (TN), total phosphorous (TP), ammonia (NH3), nitrate and nitrite (NOx), biological oxygen demand (BOD), chlorophyll-a, and hydrogen sulphide (H2S). Water samples were stored on ice in the field and then sent for testing on the same day they were sampled. Additional environmental data, including pH, dissolved oxygen (DO), and electrical conductivity (EC) were measured using a calibrated water quality unit (Horiba) at each site. The water quality unit was calibrated before and after each sampling trip.
Whole Effluent Toxicity (WET) testing
Whole Effluent Toxicity (WET) testing was conducted within the laboratory facilities of the Sydney Institute of Marine Science (SIMS) in Chowder Bay, Sydney, Australia. WET tests were completed for algal single-species and whole microbial communities in a fully-crossed experimental design created using UV-disinfected effluent from the five (5) WWTP sampled. Following standard protocols for WET testing as published by the US EPA and the ANZECC/ARMCANZ water quality guidelines (2000), five effluent concentrations were selected along with a control. The concentrations comprising 0% effluent (control), 0.1% effluent, 1% effluent, 10% effluent, 50% effluent, 90% effluent and 100% effluent.
Sample collection, preparation and testing was completed over three (3) weeks in May 2017. Disinfected effluent from each WWTP and river water samples were collected on the first day of each test week and stored in a dark constant temperature (25 °C) room O/N to allow for water temperature adjustment to the testing conditions. On each sampling occasion, a total of 30 L of disinfected effluent from each WWTP and 60 L from each river site was collected to create the dilutions for the WET tests.
Single species
The single-species WET tests were completed based on standard procedures – US EPA Test Method 1003.026 and the Environment Canada test method27 – using a 4 to 7-day old culture of the freshwater unicellular green algae Raphidocelis subcapitata (formerly also known as Selenastrum capricornutum and Pseudokirchneriella subcapitata) obtained from the NSW Department of Planning, Industry and Environment (formerly NSW Office of Environment and Heritage). Prior to WET testing, cells of R. subcapitata were washed three (3) times with artificial soft-water to remove culture media and extracellular substances. For this step, the algal culture was centrifuged at 700 g for seven (7) minutes, the supernatant discarded, and the pellet resuspended in artificial soft-water. Algal cells were counted using a haemocytometer and each test sample was inoculated with approximately 3 × 104 microalgal cells/mL.
For the single-species WET tests, 200 mL dilutions were prepared using UV-disinfected effluent (filtered at 0.45 µm) from all five (5) WWTP and artificial soft-water (filtered at 0.22 µm). Artificial soft-water used for the tests was created by the addition of sodium bicarbonate (NaHCO3), calcium sulfate dihydrate (CaSO4·2H2O), magnesium sulfate heptahydrate (MgSO4·7H20), and potassium chloride (KCl) to milli-Q water. The artificial soft-water was adjusted to the hardness of each effluent water type on the starting day of the WET tests. Total hardness as CaCO3 for effluents from each WWTP included: Kurri Kurri (106 mg/L); Farley (257 mg/L); Morpeth (78 mg/L); Raymond Terrace (65 mg/L); and Shortland (168 mg/L). Three (3) replicates of 50 mL each were added to 100 mL conical flasks and one (1) replicate of 50 mL was added to a 100 mL glass beaker. The flasks and beakers were covered with a 0.4 mm mesh to avoid any airborne particles contaminating the samples. Further, 200 µL of 2.1 g/L sodium nitrate (1.5 mg (N{O}_{3}^{-})/L) and 200 µL of 0.22 g/L potassium dihydrogen phosphate (0.15 mg (P{O}_{4}^{3-})/L) was added to each sample to provide sufficient nutrients for exponential algal growth.
The test samples were incubated for 72 hours at continuous daylight conditions. During the tests, full-spectrum daylight fluorescent lighting (36 W) provided a light intensity of 71.6 W/m2. The shelves used for the tests were lined with aluminium foil and samples were randomised once a day to maximise light exposure. At the time of inoculation (time point 0 h), the concentrations of NH3, NOx, TKN, TN, and TP, as well as water hardness were measured in each effluent type using standard inorganics and nutrients water testing suites. At the end of every nominal 24-hour period, test samples were mixed by swirling (conical flasks) or with pipette tips (beakers). 100 µL from each conical flask was taken to determine cell counts using flow cytometry, and water quality microsensors (Unisense) were used to measure pH and DO levels in the beakers.
Whole microbial community
For the community-level microbial WET tests, dilutions were prepared using unfiltered UV-disinfected effluent from all five (5) WWTP and ulfiltered river water from two (2) locations in the Hunter River estuary, including a freshwater site (salinity < 1 PSU, river site ‘F’ in Fig. 1) and a saltwater site (salinity > 30 PSU, river site ‘S’ in Fig. 1). These two (2) salinity regimes were chosen to include future scenarios based on a predicted increasing salinity of the Hunter River with rising sea levels. The community-level WET tests were run in triplicate 2 L plastic beakers for 72 hours at a 12/12 hour day/night light cycle. During the tests, LED-600 aquarium lights (BeamsWork) provided conditions suitable for the growth of photoautotrophic microbes, having a light frequency of 10,000 K (white LED) and 460 nm (blue LED), equivalent to 1,340 lumens and a light intensity of 65.1 W/m2 (assuming a luminous efficacy of 40.98 lm/W). As for the single-species tests, the shelves used for the tests were lined with aluminium foil and the beaker positions on the shelves were randomised once a day to maximise the light exposure in each beaker.
At the time of inoculation (time point 0 h), concentrations of NH3, NOx, TKN, TN, and TP were measured in each effluent type and river site sample. At the end of every nominal 24-hour period, beakers were mixed with a glass stirring rod and samples from each beaker were taken for further analysis, including 1 mL of water for microbial cell counts using flow cytometry, and approximately 500 mL of water, collected in sterile Whirl-Paks and stored on ice in the dark until filtering, for DNA extraction and sequencing. Note that filtering for DNA extraction during the WET tests followed the same protocol previously described for the microbial field surveys. Further, water quality microsensors (Unisense) were used to measure pH, DO and H2S at the sampling times (i.e. every 24 hours) for each effluent type and dilution, except Shortland WWTP due to technical difficulties. Note that DO at time point 0 h was only measured for Raymond Terrace and Morpeth effluent types due to a temporary fault in the DO sensor.
Biological oxygen demand
At the end of the community-level WET tests (time point 72 h), water was sampled from two (2) replicates of each effluent type and dilution for nutrient (NH3, NOx, TKN, TN, TP) analysis, and 200 mL of the remaining water was used to estimate the BOD of the microbial communities. The water sampled for the BOD tests were capped in standard BOD bottles and incubated for 2.5 hours in the dark. DO measurements were taken at the start of the BOD tests (T0) and after 2.5 hours incubation in the dark (T1).
Flow cytometry
Staining optimization
The nucleic acid dye SYTO9 (ThermoFisher Scientific), which has been shown to preferentially stain live and dead total bacteria cells, was tested at varying dilutions (1:80, 1:500, 1:1000 v/v of the dye’s stock solution and dimethyl sulfoxide (DMSO))28,29. All dilutions were tested using triplicate positive (stained) and negative controls (no stain). After stain optimisation using an analogue river water sample, all aliquots were subsequently stained using 0.2 µL of SYTO9 (1:1000 v/v dilution of commercial stock solution with DMSO) and incubated for 15 minutes in the dark at room temperature prior to analysis28. Note the differentiation and quantification of both live and dead cells, using Propidium Iodide, was purposely omitted, since sequencing typically incorporates both live and dead cells, and the results were aimed at determining the total number of bacteria cells (live and dead) present in the sample.
Flow cytometry analysis
All cell count samples were analysed using a LSRFortessa SORP (BD Biosciences) flow cytometer with the FACSDiva software (v8.0.1) in the Mark Wainwright Analytical Centre (Flow Cytometry Facility) at UNSW Sydney. Cell count samples from the single-species and microbial community WET tests were stored on ice until flow cytometry analysis. The samples analysed in May 2017 (‘Fresh’) were left unstained and the autofluorescence from chlorophyll-a present in the live algal cells was used to determine cell densities. The chlorophyll-a fluorescence assays were designed to compare the growth response of single-celled algae between the two (2) WET setups. Additional water samples from the WET tests were stored at −20 °C and later used to quantify the total bacterial cell densities in February and March 2019 (‘Frozen’). This was done to distinguish between the bacterial and algal (chlorophyll-a) cell populations in the water samples.
All cell count samples were separated into 200 μL aliquots across eight (8) microplates (96-wells) by effluent type (Kurri Kurri and Farley, Morpeth and Raymond Terrace) and sampling time (0 h, 24 h, 48 h, 72 h). The Frozen samples were thawed quickly (at 37 °C) to avoid cell loss during the thawing process30, and after thawing, were mixed via manual shaking for 10 s. The defrosted 200 μL aliquots in the microplate wells were stained and analysed automatically in standard-throughput mode.
A threshold of 200 cell counts was set on the side-scattered (SSC) light detector to exclude noise from non-algal and unstained bacterial cell particles. All readings were collected as logarithmic signals at a flow rate of 2.0 µL/s. Maximum events were set to 10,000,000 to ensure the counting of all cells within a 60 µL sample (100 µL for the Fresh samples). Before cell counting, all samples were mixed twice-through by mechanical pipetting up and down of 100 µL of sample at a mixing rate of 180 µL/s. Note there was no cell count data available for the community-level WET tests containing the Shortland effluent type due to complications with the flow cytometer at the time of the analysis.
Enumeration and size
The 488 nm laser was used for excitation of both SYTO9 stain and chlorophyll-a fluorescence. The detection was measured using emission filters for green (530 nm ± 15 nm) and red (780 nm ± 60 nm) fluorescence. Determination of the region within the SSC vs chlorophyll bivariate density plot, which included live algal cells containing chlorophyll-a, was done prior to the WET tests, using algal cell cultures and negative controls. Further, determination of the region defined for total bacteria within the SSC vs green fluorescence plot, was done during staining optimisation, using positive (stained) and negative (unstained) controls. Example cytograms of bacterial and chlorophyll-a populations resolved by fluorescence for 50% dilution of Kurri Kurri effluent type in freshwater (replicate 1) at 48 hours are provided in Fig. 2. Figure 2a shows the total population which is separated into quadrants within the green vs red fluorescence plot (Fig. 2b). The region defined for total bacteria is labelled “Bacteria”, while the region for cells containing chlorophyll and SYTO9 stain is labelled “Double positive”. The double positives were counted as bacteria due to the separation shown in SSC vs green florescence (SYTO9 stain) (Fig. 2d). Final cell counts were determined as the number of events within the previously selected regions. Presentation of the data as bivariate density plots (Fig. 2) enabled the best distinction between stained bacteria and chlorophyll-a. Following enumeration of the microbial cells, size was estimated using CountBright absolute counting beads (200 nm, 500 nm, 800 nm, 1 um, 3 um, 6 um) as volumetric standards. There may be some variation in the size estimates caused by the way cells are placed during the analysis.
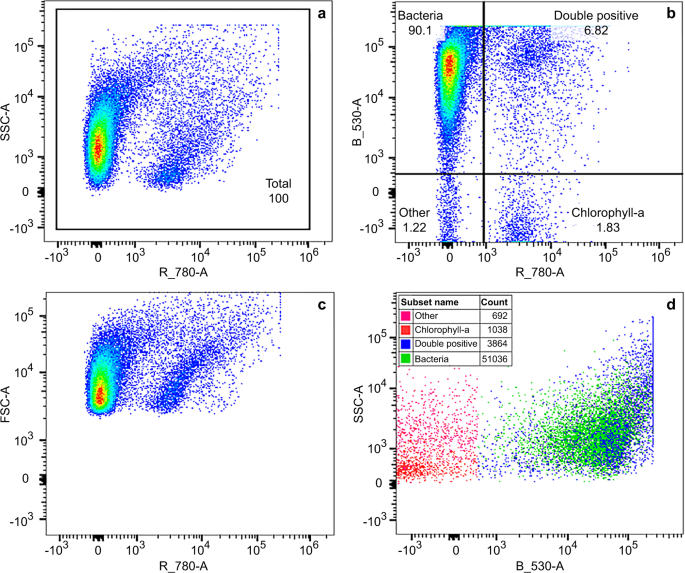
Example cytograms of bacterial and chlorophyll-a populations resolved by fluorescence for 50% dilution of Kurri Kurri effluent type in freshwater (replicate 1) at 48 hours, February and March 2019 dataset. A bivariate density plot of SSC vs red fluorescence (chlorophyll) shows the total population (a). Regions on the green vs red fluorescence plot were defined for bacteria (Bacteria), cells containing chlorophyll and SYTO9 stain (Double positive), cells containing only chlorophyll (Chlorophyll-a) and all other cells (Other) (b). Double positives were counted as total bacteria due to the separation of cells shown in SSC vs green fluorescence (d).
DNA extraction and sequencing
DNA was extracted using the DNeasy PowerWater Kit (Qiagen) following manufacturer protocols and stored at −80 °C until sequencing. Bacterial communities were identified using the 27f (5′-3′: AGAGTTTGATCMTGGCTCAG31) and 519r (5′-3′: GWATTACCGCGGCKGCTG32) primers for the V1-V3 region of the 16 S rRNA gene and Illumina™ MiSeq. 2 × 300 base pair (bp), paired-end v2 sequencing runs across two (2) lanes. Eukaryotic composition was determined using the 1391 f (5′-3′: GTACACACCGCCCGTC) and EukBr (5′-3′: TGATCCTTCTGCAGGTTCACCTAC) primer set33 for the V9 region of the 18S rRNA gene and Illumina™ MiSeq. 2 × 150 bp paired-end v3 sequencing runs across two (2) lanes. All sequencing was done at the Ramaciotti Centre for Genomics (UNSW Sydney). All raw sequence data are publicly available through the National Center for Biotechnology Information (NCBI)34 under SRA study accession SRP224901. The SRA data record includes 1,596 experiments derived from 1,476 samples.
Microfluidic quantitative polymerase chain reaction (MFQPCR)
The absolute abundance of nutrient (carbon, nitrogen, phosphorus and sulfur) cycling genes and genes associated with pathogens and antibiotic resistance were determined using microfluidic quantitative polymerase chain reaction (MFQPCR) on the Fluidigm platform. Like traditional qPCR, this method enables the determination of gene abundances through the measurement of fluorescence after each PCR cycle, but eliminates much of the manual pipetting from the sensitive qPCR reaction. Its use in ecological research in various environments has significantly increased in recent years, with a focus on fast and reliable detection of pathogens35,36,37.
Gene abundances were measured in two (2) out of three (3) replicates collected for microbial analyses from both the field surveys and WET tests. Suitable primers, as defined by38, were selected from the literature. Five (5) different gBlock gene fragments (Integrated DNA Technologies39) were designed as standards for the assays using targeted sequences for each primer pair sourced from NCBI (refer to Table 3) using Primer-BLAST40. Standard curves were generated using a dilution series of 101 to 107 copies/µL. Exact DNA concentrations of the samples were determined spectrophotometrically using the PicoGreen double-strand DNA kit (Life Technologies) on the ClarioSTAR® microplate reader (BMG Labtech) and samples were diluted to a final DNA concentration of approximately 7–8 ng/µL. To alleviate sample-specific inhibition41, DNA extracts were mixed with T4 gene 32 protein (New England Biolabs®), to a final concentration of 80 ng/uL. Specific target amplification (14 cycles) and MFQPCR were conducted at the UNSW Sydney Ramaciotti Centre for Genomics, as per 38 using Evagreen® chemistry and the 96.96 Fluidigm Dynamic Array™ Integrated Fluidic Circuit. Thermocycling consisted of 95 °C for 1 min, followed by 35 cycles of 96 °C for 5 s and 60 °C for 20 s or 25 s, followed by melt curve analysis for 60–95 °C at a ramp rate of 1 °C/3 s. Reactions were conducted in triplicate in 6.7 nL volumes with a final primer concentration of 700 nM.
Source: Resources - nature.com

