Discovery of novel candidate virus species
Two high-throughput sequencing efforts, combined with PCR and RACE, allowed us to assemble the genomes of six unique viral candidate species associated with S. solidus. BLAST searches against the genome of S. solidus did not yield significant matches, confirming that the viruses are not endogenous.
The first virus, named Schistocephalus solidus Rhabdovirus 1 (SsRV1; accession number MN803433), showed a maximum of 59% amino acid (aa) identity to the RdRP of unassigned and partially sequenced Bat Rhabdovirus (AIF4284.1) from the family Rhabdoviridae, order Mononegavirales. The SsRV1 genome encodes the five canonical proteins N-P-M-G-L with an additional short protein between G and L (Fig. 2e). All identified ORFs were flanked by conserved transcription initiation (UUGU) and transcription termination/polyadenylation sequences (UC[U]7) with very short intergenic region (Table S4). The L protein included the Mononegavirales-like RdRP domain (pfam 00946), the Mononegavirales mRNA capping region V (pfam 14318), a paramyxovirus-like mRNA capping enzyme (TIGR04198), and a Mononegavirales virus-capping methyltransferase (pfam 14314).
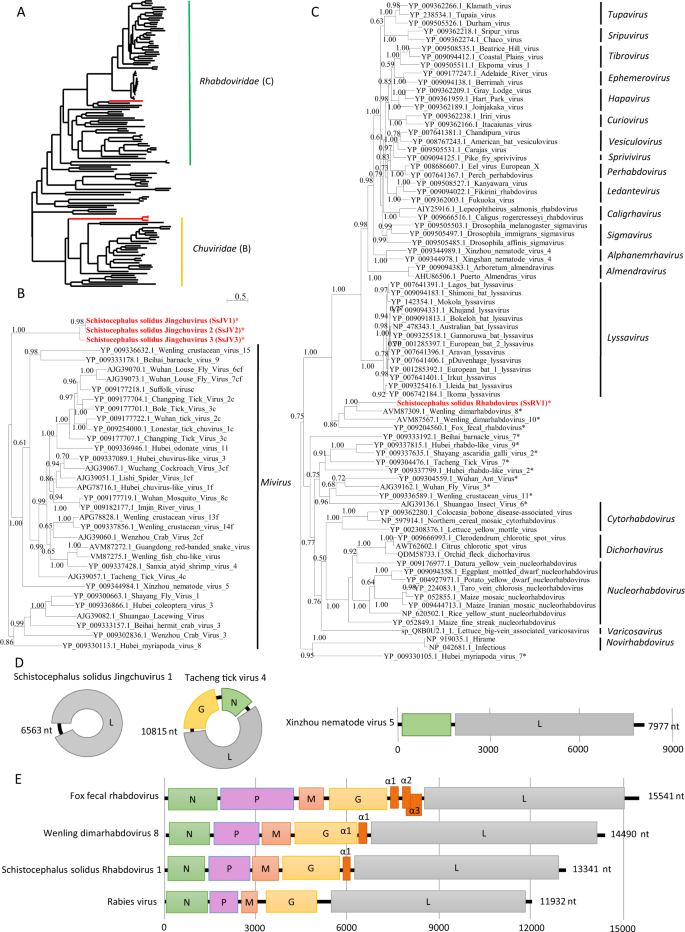
a Phylogenetic analysis of the RdRP of viruses from the order Mononegavirales and Jingchuvirales. The tree was inferred with PhyML using the LG substitution model. b, c close up views of the phylogenetic tree of the RdRP of viruses from the families Chuviridae and Rhabdoviridae, respectively. Values next to the branch indicate the results of a Shimodaira–Hasgawa branch test. Genus and family names are provided next to the branches. Asterisk (*) indicates unassigned viruses. c indicates circular genomes. f indicates fragmented genomes. d, e Genome organization of viruses from S. solidus compared with the genome of closely related viruses of the families Chuviridae and Rhabdoviridae, respectively. Boxes represent putative genes. The black line indicates noncoding regions.
The second virus, named Schistocephalus solidus Jingchuvirus 1 (SsJV1; accession number MN803434), had a circular genome encoding a single protein (Fig. 2d) with a maximum of 28% aa identity to the L protein of Hubei Myriapoda virus 8 (YP_009330113.1) of the order Jingchuvirales. The predicted protein possesses a Mononegavirales RdRP domain (pfam 00946), a paramyxovirus mRNA capping enzyme (TIGR04198) and the Mononegavirales virus-capping methyltransferase (pfam 14314).
We inferred a phylogenetic tree using the predicted RdRP sequences from SsRV1, SsJV1, and representative members of the orders Jingchuvirales and Mononegavirales (Figs. 2a and S2). Sequences clustered following established genera and families ratified by the ICTV, except for S. solidus-associated viruses, which grouped into distinct clades (Figs. 2a and S1). Our results show that SsJV1 belongs to the order Jingchuvirales, and likely represents a new taxon within the family Chuviridae (Fig. 2b). SsRV1 belongs to the family Rhabdoviridae, grouping closely with viruses reported from metatranscriptomic studies and whose host association remains unknown, including bat rhabdovirus, fox fecal rhabdovirus, Wenling dimarhabdovirus 8, and Wenling dimarhabdovirus 10 [48, 49]. Notably, SsRV1 and these closely related viruses represent a new taxon basal to Lyssavirus and to the dimarhabdovirus supergroup (Fig. 2c), with genomes of variable length, and characterized by the presence of one to three small proteins in the region between G and L (Fig. 2e).
The third viral genome, named Schistocephalus solidus bunya-like virus 1 (SsBV1; accession number MN803432), had a maximum of 36% identity to the RdRP of the Beihai barnacle virus 5 (APG79235.1). The longest predicted ORF (Fig. 3b) possesses a bunyavirus RdRP domain (pfam04196). Notably, SsBV1 was only found in sequencing data from the total RNA library and PCR assays confirmed its absence in samples that went through viral purification. We inferred a phylogenetic tree using SsBV1 and the L segment of representative members of all assigned families within the order Bunyavirales. The tree confirmed that SsBV1 has no known relatives and likely constitutes a new family of viruses (Fig. 3a).
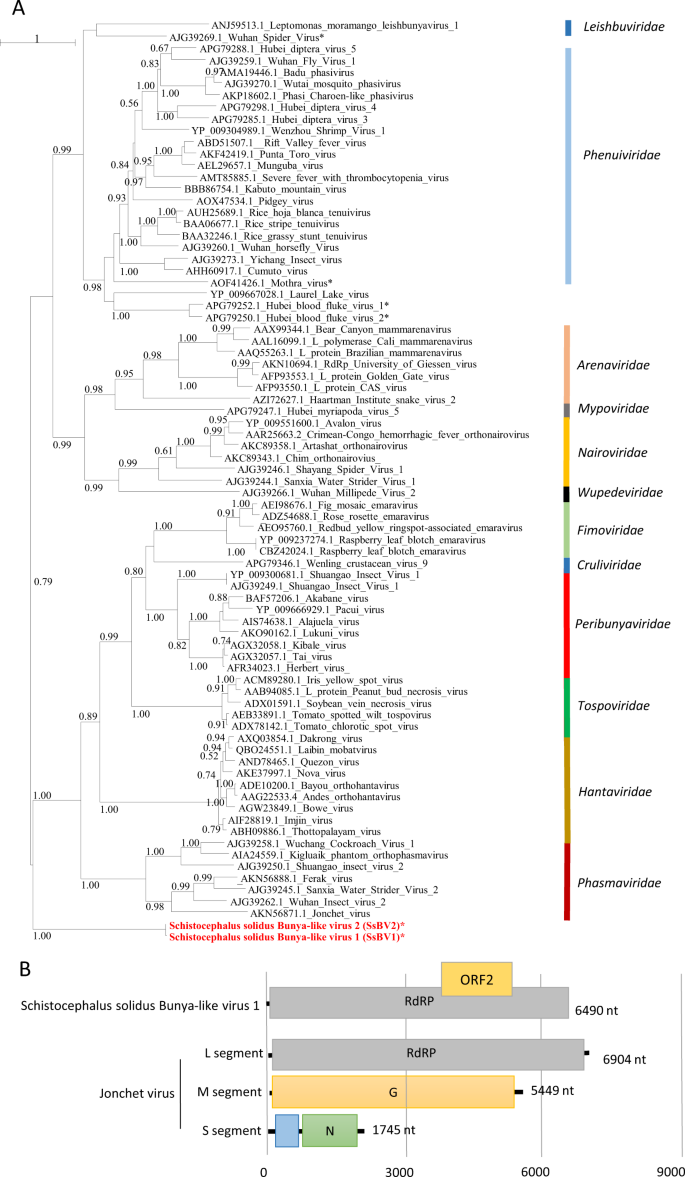
a Phylogenetic analysis of the RdRP of viruses of the order Bunyavirales. The tree was inferred with PhyML using the LG substitution model. Values next to the branch indicate the results of a Shimodaira–Hasgawa branch test. Asterisk (*) indicates unassigned viruses. Family names are provided next to the branches. b Genome organization of S. solidus bunya-like virus aligned to the genome of a related virus.
Finally, three sequences similar to dsRNA viruses of the families Totiviridae and Chrysoviridae (Fig. 4) were found and named Schistocephalus solidus toti-like viruses (SsTV1, SsTV2, and SsTV3; accession numbers MN803435 through MN803437). These viral sequences showed the highest similarities to the partial RdRP sequence of Dumyat virus (QAY29251.1, SsTV1, 27% aa identity and SsTV3, 28% aa identity), or to Hubei toti-like virus 10 (YP_009336493.1, SsTV2, 36% aa identity). All three viruses had two ORFs (Fig. 4b), with the second protein encoding for an RdRP similar to Luteovirus, Totivirus, and Rotavirus (pfam02123). We inferred a third phylogenetic tree using SsTV1, SsTV2, and SsTV3 together with 40 viruses representing the families Totiviridae, Chrysoviridae, and unassigned members closely related to these families (Fig. 4). Phylogenetic analyses revealed that SsTV1 and SsTV3 cluster together and are most closely related to viruses discovered in other invertebrates including Lophotrochozoa, Nematoda, Crustacea, and Insecta, whereas SsTV2 was most closely related to viruses discovered in insects.
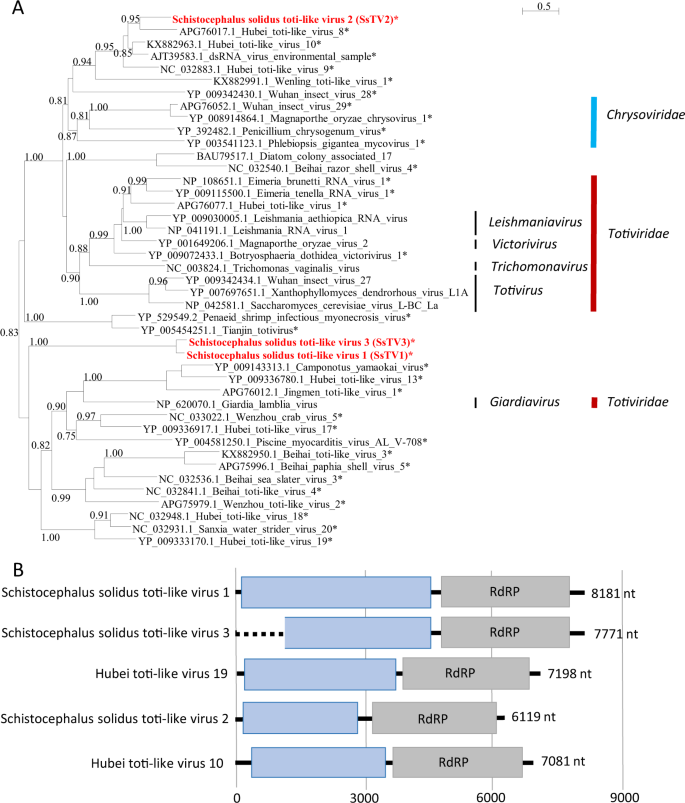
a Phylogenetic analysis of the RdRP of dsRNA viruses. The tree was inferred with PhyML using the LG substitution model. Values next to the branch indicate the results of a Shimodaira–Hasgawa branch test. Asterisk (*) indicates unassigned viruses. Genus and family names are provided next to the branches. b Genome organization of toti-like viruses from S. solidus aligned to the genome of closely related viruses.
Virus detection in geographically distant S. solidus populations through data mining
At the time of this study, two transcriptomic studies of S. solidus were publicly available. BLASTx searches against sequences from individuals from Germany and Norway ([50], PRJEB7355, two biosamples) revealed the presence of related strains of SsJV1 and SsTV1 in one dataset (ERX589070) and of SsTV2 in two datasets (ERX589070 and ERX589072). BLASTx searches against the de novo assembled transcriptome of S. solidus from Clatworthy Reservoir in Somerset, England (TSA; PRJNA304161, 15 individuals) [44] revealed two contigs representing viruses, named here SsJV2 and SsJV3, with high similarity to SsJV1 (Fig. 2). SsJV2 (GEEE01006270.1) corresponded to the full-length sequence of a variant with 94% aa sequence identity to the RdRP of SsJV1. SsJV3 (GEE01008921.1) covered only a part of the RdRP, and shared 63% aa sequence identity with SsJV1.
We further investigated the presence of viruses within the samples from England by analyzing raw sequencing reads. Viral contigs were mostly assembled from three of the 15 individuals (A12, SRR2966898, I98-2, SRR2966894, and A07, SRR2966897; Fig. S3). In addition to the above-mentioned SsJVs, we assembled a full-length sequence of the L segment of a bunya-like virus, named here SsBV2, whose genome shares 97.5% aa identity with SsBV1 (Fig. 3). Partial sequences from toti-like viruses covering 43% and 31% of the SsTV2 genome were identified and named SsTV4 and SsTV5, respectively. The consensus sequences displayed 95 and 52% aa identity to ORF1 of SsTV2. Finally, we assembled the genomes of two additional viruses, unrelated to S. solidus viruses from Alaska, which show 40.7% aa identity to the RdRP of Tapwovirus (Fig. S2). The two novel viruses, named SsNV1 and SsNV2 for Schistocephalus solidus Nyami-like virus share 71% nucleotide identity. Virus abundance varied substantially with viral reads representing between 0.0001 and 0.057% of the total number of reads. The most abundant viruses, all found in adult worms, were SsBV2, SsNV1, and SsJV2 with 85, 50, and 24 transcripts per millions of reads, respectively (Fig. S3).
Prevalence and transmission mode
We tested virus prevalence in plerocercoids from field-sampled sticklebacks from three lakes in Alaska (Figs. 5a and S4–S10). SsRV1 was the most prevalent, with an overall prevalence of 81% (±7%). In contrast, SsJV1 was detected in 10% (±6%) of plerocercoids, SsTV2 and SsBV1 were each detected in 4% (±4%) of plerocercoids, and SsTV1 and SsTV3 had a prevalence of only 2% (±2%). Only 17.5% (±7%) of plerocercoids across all populations were free of all tested viruses. A Chi-square test revealed different prevalence of SsRV1 (P < 0.01) and of virus-free individuals (P < 0.05) among populations (Figs. 5a and S3). For all viruses, we observed instances where virus(+) and virus(−) plerocercoids coinfected the same stickleback host: SsRV1 (six instances), SsJV1 (two instances), SsBV1 (one instance), SsTV1 (two instances), SsTV2 (four instances), and SsTV3 (two instances) (Figs. 5a and S4–S10).

a Virus prevalence in plerocercoids from field-collected sticklebacks and in coracidia from in vitro generated families. b SsRV1, SsJV1, and SsTV1 presence was assessed in copepods experimentally exposed and infected by S. solidus. c SsRV1 and SsJV1 presence was assessed in tissues of sticklebacks experimentally exposed and infected by S. solidus.
We tested virus presence in coracidia hatched from eggs collected upon in vitro breeding. Thirty-four families were SsRV1(+), four families were SsJV1(+), one family was SsTV1(+), one family was SsTV2(+), and one family was SsTV3(+), indicating that these viruses are vertically transmitted (Figs. 5a, S4 and S11). None of the plerocercoids used for breeding was SsBV1(+), preventing us from testing the virus transmission. To estimate the rate of vertical transmission, we experimentally infected copepods with individual coracidia hatched from virus(+) families. The presence of viruses in procercoids was assessed for 50 individuals (Fig. 5b). SsTV2(+) and SsTV3(+) families had very low hatching success, preventing us from running this experiment. SsRV1 and SsJV1 were found in all 50 tested procercoids, indicating a 100% success of vertical transmission of both viruses (Figs. S12 and S13). In contrast, 48% of the procercoids were infected by SsTV1 (Fig. S14).
Cross-species transmission to parasitized hosts
To assess the potential for S. solidus viruses to be transmitted to S. solidus intermediate and definitive hosts, we tested virus presence within the secretory products of S. solidus. We found SsRV1 in culture medium used for breeding all 12 SsRV1(+) families, whereas SsJV1, SsTV1, SsTV2, or SsTV3 were not found in media (Fig. S19).
To assess virus transmission in the first intermediate host, we tested SsRV1, SsJV1, and SsTV1 presence in copepods exposed to virus(+) parasites, but unsuccessfully infected by S. solidus. The absence of S. solidus in these copepods was confirmed by targeted PCR (Fig. S16). SsRV1 was found in exposed but noninfected copepods indicating cross-species virus transmission from S. solidus to copepods (Figs. 5b and S15). No virus was found in control nonexposed copepods. We did not test virus presence in infected copepods because we could not ensure the absence of contamination from S. solidus upon dissection.
To assess virus transmission to the second intermediate host, we tested SsRV1 and SsJV1 presence in experimentally exposed sticklebacks (Fig. 5c). Because SsTV1 success of vertical transmission was <100%, this virus was excluded from the experiment. No virus was found in tissues of sticklebacks exposed but noninfected by S. solidus (four tested individuals). Four sticklebacks successfully infected by an SsRV1(+) parasite carried the virus within their liver, spleen, and head kidney, but the virus was absent from the fish gut (Fig. S17). The one fish that was successfully infected by an SsJV1(+) parasite did not transmit the virus to its fish host (Fig. S17).
To further assess virus transmission to parasitized sticklebacks, we tested virus presence in the liver of field-samples sticklebacks. SsRV1 was found in the liver of the 19 sticklebacks parasitized by SsRV1(+) parasites but it was absent from the 5 sticklebacks parasitized by SsRV1(−) parasites. Six sticklebacks were coinfected by both SsRV1(+) and SsRV1(−) parasites, and SsRV1 was found in the liver of all sticklebacks parasitized by at least one SsRV1(+) parasite (Fig. S18). None of the other viruses was found in the liver of field-sampled sticklebacks.
Source: Ecology - nature.com
