Bacterial strains and culture conditions
The bacterial strains used in this study are listed in Supplementary Table 2. Bacterial cultures were grown at 37 °C from frozen stocks on Luria-Bertani (LB: 1% tryptone (Oxoid), 0.5% yeast extract (Oxoid) and 0.5% NaCl) plates. Isolated bacteria were inoculated in the appropriate medium. The biotrophic fungus Podosphaera xanthii was grown at 25 °C from a frozen stock on cucumber cotyledons and maintained on them until inoculum preparation. Biofilm assays were performed on MSgg medium: 100 mM morpholinepropane sulfonic acid (MOPS) (pH 7), 0.5% glycerol, 0.5% glutamate, 5 mM potassium phosphate (pH 7), 50 μg/ml tryptophan, 50 μg/ml phenylalanine, 50 μg/ml threonine, 2 mM MgCl2, 700 μM CaCl2, 50 μM FeCl3, 50 μM MnCl2, 2 μM thiamine, 1 μM ZnCl2. For the in vitro lipopeptide detection and assays with cell-free supernatants, medium optimized for lipopeptide production (MOLP)98 was used: 30 g/liter peptone, 20 g/liter saccharose, 7 g/liter yeast extract, 1.9 g/liter KH2PO4, 0.001 mg/ liter CuSO4, 0.005 mg/liter FeCl3.6H2O, 0.004 mg/liter Na2MoO4, 0.002 mg/liter KI, 3.6 mg/liter MnSO4.H2O, 0.45 g/liter MgSO4, 0.14 mg/liter ZnSO4.7H2O, 0.01 mg/liter H3BO3, and 10 mg/liter citric acid. The pH was adjusted to 7 with 5 M NaOH prior to sterilization. For cloning and plasmid replication, Escherichia coli DH5α was used. Escherichia coli BL21(DE3) was used for protein purification. Bacillus subtilis 168 is a domesticated strain used to transform the different constructs into Bacillus subtilis NCIB3610. The antibiotic final concentrations for B. subtilis were: MLS (1 μg/ml erythromycin, 25 μg/ml lincomycin); spectinomycin (100 μg/ml); tetracycline (10 μg/ml); chloramphenicol (5 μg/ml); and kanamycin (10 μg/ml).
Strain construction
All of the primers used to generate the different strains are listed in Supplementary Table 3. To build the strain YNG001, the promoter of the fengycin operon was amplified with the Ppps-ecoRI.F and Ppps-HindIII.R primer pair. The PCR product was digested with EcoRI and HindIII and cloned into the pKM003 vector cut with the same enzymes. The resulting plasmid was transformed by natural competence into B. subtilis 168 replacing the amyE neutral locus. Transformants were selected via spectinomycin resistance. The same plasmid was used to build the strain YNG002 by transforming a ΔtasA strain of B. subtilis 168.
Strain YNG003 was constructed using the primers amyEUP-Fw, amyEUP-Rv, Ppps-Fw, Ppps-Rv, Yfp-Fw, Yfp-Rv, Cat-Fw. Cat-Rv, amyEDOWN-Fw, and amyEDOWN-Rv to separately amplify the relevant fragments. The fragments were then joined using the NEB builder HiFi DNA Assembly Master Mix (New England Biolabs). The construct was made using pUC19 digested with BamHI as the vector backbone. The final plasmid was then transformed into B. subtilis 168 replacing amyE, and transformants were selected via chloramphenicol resistance.
Strain JC97 was generated using the primers bslAUP-Fw, bslADOWN-Rv, Spc-Fw, Spc-Rv, bslaUP-Fw and bslADOWN-Rv, and XbaI-digested pUC19 as the vector backbone. The fragments were assembled using NEB Builder HiFi DNA Assembly Master Mix.
Strains JC70, JC81, and JC149 were constructed via site-directed mutagenesis (QuickChange Lightning Site Directed Mutagenesis Kit – Agilent Technologies). Briefly, the tapA operon (tapA-sipW-tasA), including its promoter, was amplified using the primers TasA_1_mutb and YSRI_2, and the resulting product was digested with BamHI and SalI and cloned into the pDR183 vector99. Next, the corresponding primers (Supplementary Table 3) were used to introduce the alanine substitution mutations into the desired positions of the TasA amino acid sequence. The entire plasmid was amplified from the position of the primers using Pfu DNA polymerase. The native plasmid, which was methylated and lacked the mutations, was digested with DpnI enzyme. The plasmids containing the native version of TasA (JC70) or the mutated versions (JC81 and JC149) were transformed into the B. subtilis 168 Δ(tapA-sipW-tasA) strain replacing the lacA neutral locus. Genetic complementation was observed in strain JC70 as a control. Transformants were selected via MLS resistance.
Plasmid pDFR6 (pET22b-tasA), which contains the open reading frame of the tasA gene from B. subtilis NCIB3610 without the signal peptide or the stop codon, was constructed as previously described76.
Primers used in the analysis of gene expression by qRT-PCR are listed in Supplementary Table 4.
All of the B. subtilis strains generated were constructed by transforming B. subtilis 168 via its natural competence and then using the positive clones as donors for transferring the constructs into B. subtilis NCIB3610 via generalized SPP1 phage transduction100.
Biofilm assays
To analyze colony morphology under biofilm-inducing conditions101, the bacterial strains were grown on LB plates overnight at 37 °C, and the resulting colonies were resuspended in sterile distilled water at an OD600 of 1. Next, 2-µl drops of the different bacterial suspensions were spotted on MSgg or LB agar (depending on the assay) agar plates and incubated at 30 °C. Colonies were removed at the appropriate time points (24, 48, and 72 h) for the different analyses.
For the Δeps-ΔtasA co-inoculation assay, colonies were resuspended in sterile distilled water and mixed at a final OD600 of 1. Next, the bacterial suspension was inoculated onto MSgg agar plates and incubated as described above. For the external complementation assay using purified TasA, a drop containing 80 µg of protein was spotted onto MSgg agar plates and allowed to dry. Next, ΔtasA cells were inoculated on top of the dried drop and incubated as described above.
For the CFU counts of the colonies from the different strains, 24-, 48- and 72-h-old colonies grown on MSgg agar plates were removed, resuspended in 1 ml of sterile distilled water, and subjected to mild sonication (three rounds of 20 second pulses at 20% amplitude). The resulting suspensions were serially diluted and plated to calculate the CFUs per colony (total CFU). To estimate the CFUs corresponding to sporulated cells (CFU endospores), the same dilutions were heated at 80 °C for 10 min and plated. The sporulation percentage was calculated as (CFU endospores/total CFU) * 100.
Biofilm fractionation
To analyze the presence of TasA in the different strains, biofilms were fractionated into cells and ECM101. Both fractions were analyzed separately. In all, 72-h-old colonies grown under biofilm-inducing conditions on MSgg-agar plates were carefully lifted from the plates and resuspended in 10 ml of MS medium (MSgg broth without glycerol and glutamate, which were replaced by water) with a 25 5/8 G needle. Next, the samples were subjected to mild sonication in a Branson 450 digital sonifier (4–5 5 s pulses at 20% amplitude) to ensure bacterial resuspension. The bacterial suspensions were centrifuged at 9000 × g for 20 min to separate the cells from the extracellular matrix. The cell fraction was resuspended in 10 ml of MS medium and stored at 4 °C until further processing. The ECM fraction was filtered through a 0.22-µm filter and stored at 4 °C.
For protein precipitation, 2 ml of the cell or ECM fractions were used. The cell fraction was treated with 0.1 mg/ml lysozyme for 30 min at 37 °C. Next, both fractions were treated with a 10% final concentration of trichloroacetic acid and incubated in ice for 1 h. Proteins were collected by centrifugation at 13,000 × g for 20 min, washed twice with ice-cold acetone, and dried in an Eppendorf Concentrator Plus 5305 (Eppendorf).
Cell membrane fractionation
Crude membrane extracts were purified from 50 ml MSgg liquid cultures (with shaking) of the different B. subtilis strains. Cultures were centrifuged at 7000 × g for 10 min at 4 °C and then resuspended in 10 ml of PBS. Lysozyme was added at a final concentration of 20 µg/ml and the cell suspensions were incubated at 37 °C for 30 min. After incubation, the lysates were sonicated on ice with a Branson 450 digital sonifier using a cell disruptor tip and 45 s pulses at 50% amplitude with pauses of 30 s between pulses until the lysates were clear. Next, the cell lysates were centrifuged at 10,000 × g for 15 min to eliminate cell debris, and the supernatants were separated and passed through a 0.45-µm filter. To isolate the cell membrane, the filtered lysate was ultracentrifuged at 100,000 × g for 1 h at 4 °C. The supernatant, which contained the cytosolic proteins, was separated and kept at −20 °C. The pellet, which contained the crude membrane extract, was washed three times with PBS and processed using the CelLytic MEM protein extraction kit from Sigma. Briefly, the membrane fractions were resuspended in 600 µl of lysis and separation working solution (lysis and separation buffer + protease inhibitor cocktail) until a homogeneous suspension was achieved. Next, the suspension was incubated overnight at 4 °C on a stirring wheel. After incubation, the suspension is incubated at 37 °C for 30 min and then centrifuged at 3000 × g for 3 min. The DSM (upper phase) was separated and kept at −20 °C, and the DRM (lower phase) was washed three times with 400 µl of wash buffer by repeating the process from the 37 °C incubation step. Three washes were performed to ensure the removal of all hydrophilic proteins. The isolated DRM was kept at −20 °C until use. The DRM, DSM, and cytosolic fractions were used directly for immunodetection.
Protein expression and purification
Protein was expressed and purified as previously described102 with some changes. Briefly, freshly transformed BL21(DE3) E. coli colonies were picked, resuspended in 10 mL of liquid LB with 100 µg/mL of ampicillin and incubated O/N at 37 °C with shaking. The next day, the pre-inoculum was used to inoculate 500 mL of LB supplemented with ampicillin, and the culture was incubated at 37 °C until an OD600 of 0.7–0.8 was reached. Next, the culture was induced with 1-mM isopropyl β-d-1-thiogalactopyranoside (IPTG) and incubated O/N at 30 °C with shaking to induce the formation of inclusion bodies. The next day, cells were harvested via centrifugation (5000 × g, 15 min, 4 °C) resuspended in buffer A (Tris 50 mM, 150 mM NaCl, pH8), and then centrifuged again. The pellets were kept at −80 °C until purification or processed after 15 min. After thawing, cells were resuspended in buffer A, sonicated on ice (3 × 45 s, 60% amplitude) and centrifuged (15,000 × g, 60 min, 4 °C). The supernatant was discarded, as proteins were mainly expressed in inclusion bodies. The pellet was resuspended in buffer A supplemented with 2 % Triton X-100, incubated at 37 °C with shaking for 20 min and centrifuged (15,000 × g, 10 min, 4 °C). The pellet was extensively washed with buffer A, centrifuged (15,000 × g for 10 min, 4 °C), resuspended in denaturing buffer (Tris 50 mM NaCl 500 mM, 6 M GuHCl), and incubated at 60 °C overnight until complete solubilization occured. Lysates were clarified via sonication on ice (3 × 45 s, 60% amplitude) and centrifugation (15,000 × g, 1 h, 16 °C) and were then passed through a 0.45-µm filter prior to affinity chromatography. Protein was purified using an AKTA Start FPLC system (GE Healthcare). Soluble inclusion bodies were loaded into a HisTrap HP 5 mL column (GE Healthcare) previously equilibrated with binding buffer (50 mM Tris, 0.5 M NaCl, 20 mM imidazole, 8 M urea, pH 8). Protein was eluted from the column with elution buffer (50 mM Tris, 0.5 M NaCl, 500 mM imidazole, 8 M urea, pH 8). After the affinity chromatography step, the purified protein was loaded into a HiPrep 26/10 desalting column (GE Healthcare), and the buffer was exchanged to Tris 20 mM, NaCl 50 mM to perform the corresponding experiments.
SDS-PAGE and immunodetection
Precipitated proteins were resuspended in 1x Laemmli sample buffer (BioRad) and heated at 100 °C for 5 min. Proteins were separated via SDS-PAGE in 12% acrylamide gels and then transferred onto PVDF membranes using the Trans-Blot Turbo Transfer System (BioRad) and PVDF transfer packs (BioRad). For immunodetection of TasA, the membranes were probed with anti-TasA antibody (rabbit) used at a 1:20,000 dilution in Pierce Protein-Free (TBS) blocking buffer (ThermoFisher). For immunodetection of FloT-YFP, a commercial anti-GFP primary antibody (Clontech living colors full-length polyclonal antibody) developed in rabbit were used at a 1:1000 or dilution in the buffer mentioned above. A secondary anti-rabbit IgG antibody conjugated to horseradish peroxidase (BioRad) was used at a 1:3000 dilution in the same buffer. The membranes were developed using the Pierce ECL Western Blotting Substrate (ThermoFisher).
Mass spectrometry analysis of protein bands
The sequence corresponding to the band of the ECM fraction of JC81 (Supplementary Fig. 13A) was identified via tandem mass spectrometry using a “nano” ion trap system (HPLC-ESI-MS/MS). Briefly, the bands obtained after electrophoresis were cut out, washed, and destained. Subsequently, the disulfide bridges were reduced with DTT, cysteines were alkylated via the use of iodoacetamide, and in-gel trypsin digestion was performed to extract the peptides corresponding to the protein samples. This entire process was carried out automatically using an automatic digester (DigestPro, Intavis Bioanalytical Instruments). The peptides were then concentrated and desalted using a capture column C18 ZORBAX 300SB-C18 (Agilent Technologies, Germany), 5 × 0.3 mm, with 5-µm particle diameter and 300-Å pore size, using a gradient of 98% H2O:2% acetonitrile (ACN)/0.1% formic acid (FA) with a flow rate of 20 μL/min for 6 min. The capture column was connected in line to a ZORBAX 300SB-C18 analytical column (Agilent Technologies), 150 × 0.075 mm, with a 3.5-µm particle diameter and 300-Å pore size, through a 6-port valve. Elution of the samples from the capture column was performed over a gradient using FA 0.1% in water as the mobile phase A and FA 0.1% in ACN 80%/water 20% as the mobile phase B. The LC system was coupled through a nanospray source (CaptiveSpray, Bruker Daltonics) to a 3D ion trap mass spectrometer (amaZon speed ETD, Bruker Daltonics) operating in positive mode with a capillary voltage set to 1500 V and a sweep range: m/z 300–1500. “Data-dependent” acquisition was carried out in automatic mode, which allowed the sequential collection of an MS spectrum in “full scan” (m/z 300_1400) followed by an MS spectrum in tandem via CID of the eight most abundant ions. For identification, the software ProteinScape 3 (Bruker Daltonics) coupled to the search engine Mascot 3.1 (Matrix Science) was used, matching the MS/MS data against the Swiss-Prot and NCBInr databases.
Bioassays on melon leaves
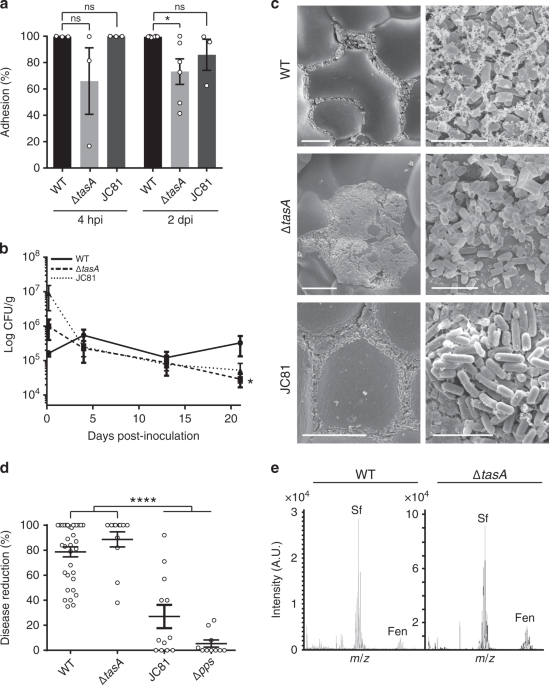
Bacterial strains were grown in liquid LB at 30 °C overnight. The cells in the cultures were washed twice with sterile distilled water. The bacterial cell suspensions were adjusted to the same OD600 and sprayed onto leaves of 4- to 5-week-old melon plants. Two hours later, a suspension of P. xanthii conidia was sprayed onto each leaf at a concentration of 4−10 × 104 spores/ml. The plants were placed in a greenhouse or in a growth chamber at 25 °C with a 16-h photoperiod, 3800 lux, and 85% RH. The severity of the symptoms in melon leaves was evaluated by the estimation of disease severity103. Disease severity was calculated by quantifying the leaf area covered by powdery mildew using FiJi104 image software analysis and pictures of infected leaves. Briefly, the channels of the image were split and the area covered by powdery mildew was measured in 8-bit images by selecting the powdery mildew damage area (white powdery stains that cover the leaf) through image thresholding, given that the stains caused by the disease have higher pixel intensity values. Total leaf area was determined by manually selecting the leaf outline using the polygon selection tool the ratio of infection was calculated using the formula (see Eq. 1):
$${mathrm{Ratio}},{mathrm{of}},{mathrm{infection}} = frac{{{mathrm{damaged}},{mathrm{area}}}}{{{mathrm{total}},{mathrm{leaf}},{mathrm{area}}}} times 100$$
(1)
The persistence of bacterial strains on plant leaves was calculated via CFU counts performed over the twenty-one days following inoculation. Three different leaves from three different plants were individually placed into sterile plastic stomacher bags and homogenized in a lab blender (Colworth Stomacher-400, Seward, London, UK) for 3 min in 10 ml of sterile distilled water. The leaf extracts were serially diluted and plated to calculate the CFUs at each time point. The plates were incubated at 37 °C for 24 h before counting.
The adhesion of bacterial cells to melon leaves was estimated by comparing the number of cells released from the leaf versus the cells attached to the surface. The surfaces of individual leaves were placed in contact with 100 ml of sterile distilled water in glass beakers and, after 10 min of stirring (300 rpm), the water and leaf were plated separately. The leaves were processed as described above. Adhesion was calculated as the ratio: (water CFU/total CFU) × 100. The data from all of the different strains were normalized to the result of the WT strain (100% adhesion).
Antifungal activity of cell-free supernatant against P. xanthii
B. subtilis strains were grown for 72 h at 30 °C in MOLP medium, and the supernatant was centrifuged and filtered (0.22 µm). One-week-old cotyledons were disinfected with 20% commercial bleach for 30 s and then submerged two times in sterile distilled water for 2 min and then air dried. 10-mm disks were excised with a sterilized cork borer, incubated with cell-free supernatants for 2 h, and then left to dry. Finally, the disks were inoculated with P. xanthii conidia on their adaxial surface with a soft paintbrush105.
Lipopeptides production analysis
For the in vitro lipopeptide detection, bacteria were grown in MOLP for 72 h. The cultures were centrifuged, and the supernatants were filtered (0.22 µm) prior to analysis via MALDI-TOF/TOF mass spectrometry.
For the analysis of lipopeptide production in colonies, WT or ΔtasA colonies were grown on MSgg plates for 72 h at 30 °C. For the cell fractions, whole colonies were resuspended as described above in 1 mL of sterile distilled water and centrifuged at 5000 × g for 5 min. The pellets were then resuspended in 1 ml of methanol and sonicated in a bath for 10 min. Cells were harvested via centrifugation at 5000 × g for 5 min, and the supernatant containing the solubilized lipopeptides was filtered through a 0.22-µm filter and stored at 4 °C prior to analysis. For the agar fraction, after the colonies were removed, a piece of agar of approximately the same surface was sliced out and introduced into a 2-mL Eppendorf tube containing glass beads. In all, 1 mL of methanol was added, and then the tube was vigorously vortexed until the agar was broken down. Finally, the mixture was sonicated in a bath for 10 min and centrifuged at 5000 × g for 5 min. The supernatant was filtered through a 0.22-µm filter and stored at 4 °C prior to analysis by MALDI-TOF/TOF.
For in situ lipopeptide detection on inoculated leaves, leaf disks were taken 21 days post-inoculation with a sterile cork borer and then placed directly on an UltrafleXtreme MALDI plate. A matrix consisting of a combination of CHCA (α-cyano-4-hydroxycinnamic acid) and DHB (2,5-dihydroxybenzoic acid) was deposited over the disks or the supernatants (for the in vitro cultures or the colonies’ analysis), and the plates were inserted into an UltrafleXtreme MALDI-TOF/TOF mass spectrometer. The mass spectra were acquired using the Bruker Daltonics FlexControl software and were processed using Bruker Daltonics FlexAnalysis.
Electron microscopy analysis
For the scanning electron microscopy analysis, leaf disks were taken 21 days post-inoculation as previously described and fixed in 0.1 M sodium cacodylate and 2% glutaraldehyde overnight at 4 °C. Three washes were performed with 0.1 M sodium cacodylate and 0.1 M sucrose followed by ethanol dehydration in a series of ethanol solutions from 50% to 100%. A final drying with hexamethyldisilazane was performed as indicated106. The dried samples were coated with a thin layer of iridium using an Emitech K575x turbo sputtering coater before viewing in a Helios Nanolab 650 Scanning Electron Microscope and Focus Ion Beam (SEM-FIB) with a Schottky-type field emission electron gun.
For the transmission electron microscopy analysis, bacterial colonies grown on MSgg agar for the appropriate times were fixed directly using a 2% paraformaldehyde-2.5% glutaraldehyde-0.2 M sucrose mix in phosphate buffer 0.1 M (PB) overnight at 4 °C. After three washes in PB, portions were excised from each colony and then post-fixed with 1% osmium tetroxide solution in PB for 90 min at room temperature, followed by PB washes, and 15 min of stepwise dehydration in an ethanol series (30%, 50%, 70%, 90%, and 100% twice). Between the 50% and 70% steps, colonies were incubated in-bloc in 2% uranyl acetate solution in 50% ethanol at 4 °C, overnight. Following dehydration, the samples were gradually embedded in low-viscosity Spurr’s resin: resin:ethanol, 1:1, 4 h; resin:ethanol, 3:1, 4 h; and pure resin, overnight. The sample blocks were embedded in capsule molds containing pure resin for 72 h at 70 °C.
For the immunolabeling assays, samples from the corresponding strains were grown under biofilm-inducing conditions at 30 °C. After 48 h of incubation, carbon-coated copper grids were deposited into the wells over the pellicles formed at the interface between the medium and the air (in the case of mutants unable to form a pellicle, copper grids were deposited in the interface) and incubated with the samples at 28 °C for 2 h. After incubation, the grids were washed in PSB for 5 min, and then the samples were fixed with a solution of 2% paraformaldehyde for 10 min, washed in PBS and blocked with Pierce Protein-Free (TBS) blocking buffer (ThermoFisher) for 30 min. Anti-TasA primary antibody was used at a 1:150 dilution in blocking buffer, and grids were deposited over drops of the antibody solution and incubated for 1 h at room temperature. Samples were washed three times with TBS -T (50 mM Tris-HCl, 150 mM NaCl, pH 7.5 – Tween20 0.1%) for 5 min and then exposed to 10-nm diameter immunogold-conjugated secondary antibody (Ted Pella) for 1 h at a 1:50 dilution. The samples were then washed twice with TBS-T and once with water for 5 min each. Finally, the grids were treated with glutaraldehyde (2%) for 10 min, washed in water for 5 min, negatively stained with uranyl acetate (1%) for 20 s and, lastly, washed once with water for 30 s.
The samples were left to dry and were visualized under a FEI Tecnai G2 20 TWIN Transmission Electron Microscope at an accelerating voltage of 80 KV. The images were taken using a side-mounted CCD Olympus Veleta with 2k x 2k Mp.
Whole-transcriptome analysis and qRT-PCR
Biofilms were grown on MSgg agar as described above. 24-, 48-, and 72-h colonies of the corresponding strains (WT or ΔtasA) were recovered and stored at −80 °C. All of the assays were performed in duplicate. The collected cells were resuspended and homogenized via passage through a 255/8 G needle in BirnBoim A107 buffer (20% sucrose, 10 mM Tris-HCl pH 8, 10 mM EDTA and 50 mM NaCl). Lysozyme (10 mg/ml) was added, and the mixture was incubated for 30 min at 37 °C. After disruption, the suspensions were centrifuged, and the pellets were resuspended in Trizol reagent (Invitrogen). Total RNA extraction was performed as instructed by the manufacturer. DNA removal was carried out via in-column treatment with the rDNAse included in the Nucleo-Spin RNA Plant Kit (Macherey-Nagel) following the instructions of the manufacturer. The integrity and quality of the total RNA was assessed with an Agilent 2100 Bioanalyzer (Agilent Technologies) and by gel electrophoresis.
To perform the RNA sequencing analysis, rRNA removal was performed using the RiboZero rRNA removal (bacteria) Kit from Illumina, and 100-bp single-end read libraries were prepared using the TruSeq Stranded Total RNA Kit (Illumina). The libraries were sequenced using a NextSeq550 instrument (Illumina). The raw reads were pre-processed with SeqTrimNext108 using the specific NGS technology configuration parameters. This pre-processing removes low quality, ambiguous and low complexity stretches, linkers, adapters, vector fragments, and contaminated sequences while keeping the longest informative parts of the reads. SeqTrimNext also discarded sequences below 25 bp. Subsequently, clean reads were aligned and annotated using the B. subtilis subsp. subtilis str. 168 genome (NC_000964.3) as the reference with Bowtie2109 in BAM files, which were then sorted and indexed using SAMtools v1.484110. Uniquely localized reads were used to calculate the read number value for each gene via Sam2counts (https://github.com/vsbuffalo/sam2counts). Differentially expressed genes (DEGs) between WT and ΔtasA were analyzed via DEgenes Hunter111, which provides a combined p value calculated (based on Fisher’s method112) using the nominal p values provided by from edgeR113 and DEseq2114. This combined p value was adjusted using the Benjamini-Hochberg (BH) procedure (false discovery rate approach)115 and used to rank all the obtained differentially expressed genes. For each gene, combined p value < 0.05 and log2-fold change >1 or <−1 were considered as the significance threshold. Heatmap and DEGs clusterization was performed using ComplexHeatmap116 in Rstudio. STEM117 was used to model temporal expression profiles independent of the data. Only profiles with a p value < 0.05 were considered in this study. The DEGs annotated with the B. subtilis subsp. subtilis str. 168 genome were used to identify the Gene Ontology functional categories using sma3s118 and TopGo Software119. Gephi software (https://gephi.org) was used to generate the DEG networks, and the regulon list was downloaded from subtiwiki (http://subtiwiki.uni-goettingen.de). The data were deposited in the GEO database (GEO accession GSE124307).
Quantitative real-time (qRT)-PCR was performed using the iCycler-iQ system and the iQ SYBR Green Supermix Kit from Bio-Rad. The primer pairs used to amplify the target genes were designed using the Primer3 software (http://bioinfo.ut.ee/primer3/) and Beacon designer (http://www.premierbiosoft.com/qOligo/Oligo.jsp?PID=1), maintaining the parameters described elsewhere120. For the qRT-PCR assays, the RNA concentration was adjusted to 100 ng/µl. Next, 1 µg of DNA-free total RNA was retro-transcribed into cDNA using the SuperScript III reverse transcriptase (Invitrogen) and random hexamers in a final reaction volume of 20 µl according to the instructions provided by the manufacturer. The qRT-PCR cycle was: 95 °C for 3 min, followed by PCR amplification using a 40-cycle amplification program (95 °C for 20 s, 56 °C for 30 s, and 72 °C for 30 s), followed by a third step of 95 °C for 30 s. To evaluate the melting curve, 40 additional cycles of 15 s each starting at 75 °C with stepwise temperature increases of 0.5 °C per cycle were performed. To normalize the data, the rpsJ gene, encoding the 30S ribosomal protein S10, was used as a reference gene121. The target genes fenD, encoding fengycin synthetase D, alsS, encoding acetolactate synthase, albE, encoding bacteriocin subtilosin biosynthesis protein AlbE, bacB, encoding the bacilysin biosynthesis protein BacB, and srfAA encoding surfactin synthetase A, were amplified using the primer pairs given in Supplementary Table 4, resulting in the generation of fragments of 147 bp, 82 bp, 185 bp, 160 bp, and 94 bp, respectively. The primer efficiency tests and confirmation of the specificity of the amplification reactions were performed as previously described122. The relative transcript abundance was estimated using the ΔΔ cyclethreshold (Ct) method123. Transcriptional data of the target genes was normalized to the rpsJ gene and shown as the fold-changes in the expression levels of the target genes in each B. subtilis mutant strain compared to those in the WT strain. The relative expression ratios were calculated as the difference between the qPCR threshold cycles (Ct) of the target gene and the Ct of the rpsJ gene (ΔCt = Ctrgene of interest – CtrpsJ). Fold-change values were calculated as 2−ΔΔCt, assuming that one PCR cycle represents a two-fold difference in template abundance124,125. The qRT-PCR analyses were performed three times (technical replicates) using three independent RNA isolations (biological replicates).
Flow cytometry assays
Cells were grown on MSgg agar at 30 °C. At different time points, colonies were recovered in 500 μL of PBS and resuspended with a 255/8 G needle. For the promoter expression assays, colonies were gently sonicated as described above to ensure complete resuspension, and the cells were fixed in 4% paraformaldehyde in PBS and washed three times in PBS. To evaluate the physiological status of the different B. subtilis strains, cells were stained without fixation for 30 min with 5 mM 5-cyano-2,3-ditolyltetrazolium chloride (CTC) and 15 µM 3-(p-hydroxyphenyl) fluorescein (HPF).
The flow cytometry runs were performed with 200 μl of cell suspensions in 800 μL of GTE buffer (50 mM glucose, 10 mM EDTA, 20 mM Tris-HCl; pH 8), and the cells were measured on a Beckman Coulter Gallios™ flow cytometer using 488 nm excitation. YFP and HPF fluorescence were detected with 550 SP or 525/40 BP filters. CTC fluorescence was detected with 730 SP and 695/30BP filters. The data were collected using Gallios™ Software v1.2 and further analyzed using Kaluza Analysis v1.3 and Flowing Software v2.5.1. Negative controls corresponding to unstained bacterial cells (or unlabeled cells corresponding to each strain for the promoter expression analysis) were used to discriminate the populations of stained bacteria in the relevant experiments and for each dye (Supplementary Fig. 19).
Intracellular pH analysis
Intracellular pH was measured as previously described55. Colonies of the different strains grown on MSgg agar at 30 °C were taken at different time points and recovered in potassium phosphate buffer (PPB) pH 7 and gently sonicated as described above. Next, the cells were incubated in 10 µl of 1 mM 5-(6)carboxyfluorescein diacetate succinimidyl (CFDA) for 15 min at 30 °C. PPB supplemented with glucose (10 mM) was added to the cells for 15 min at 30 °C to remove the excess dye. After two washes with the same buffer, the cells were resuspended in 50 mM PPB (pH 4.5).
Fluorescence was measured in a FLUOstar Omega (BMG labtech) microplate spectrofluorometer using 490 nm/525 nm as the excitation and emission wavelengths, respectively. Conversion from the fluorescence arbitrary units into pH units was performed using a standard calibration curve.
Confocal laser scanning microscopy
Cell death in the bacterial colonies was evaluated using the LIVE/DEAD BacLight Bacterial Viability Kit (Invitrogen). Equal volumes of both components included in the kit were mixed, and 2 µl of this solution was used to stain 1 ml of the corresponding bacterial suspension. Sequential acquisitions were configured to visualize the live or dead bacteria in the samples. Acquisitions with excitation at 488 nm and emission recorded from 499 to 554 nm were used to capture the images from live bacteria, followed by a second acquisition with excitation at 561 nm and emission recorded from 592 to 688 nm for dead bacteria.
For the microscopic analysis and quantification of lipid peroxidation in live bacterial samples, we used the image-iT Lipid Peroxidation Kit (Invitrogen) following the manufacturer’s instructions with some slight modifications. Briefly, colonies of the different strains were grown on MSgg plates at 30 °C, isolated at different time points, and resuspended in 1 ml of liquid MSgg medium as described in the previous sections. In all, 5 mM cumene hydroperoxide (CuHpx)-treated cell suspensions of the different strains at the corresponding times were used as controls. The cell suspensions were then incubated at 30 °C for 2 h and then stained with a 10-µM solution of the imageIT lipid peroxidation sensor for 30 min. Finally, the cells were washed three times with PBS, mounted, and visualized immediately. Images of the stained bacteria were acquired sequentially to obtain images from the oxidized to the reduced states of the dye. The first image (oxidized channel) was acquired by exciting the sensor at 488 nm and recording the emissions from 509 to 561 nm, followed by a second acquisition (reduced channel) with excitement at 561 nm and recording of the emissions from 590 to 613 nm.
Membrane potential was evaluated using the image-iT TMRM (tetramethylrhodamine, methyl ester) reagent (Invitrogen) following the manufacturer’s instructions. Colonies grown at 30 °C on MSgg solid medium were isolated at different time points and resuspended as described above. Samples treated prior to staining with 20 µM carbonyl cyanide m-chlorophenyl hydrazine (CCCP), a known protonophore and uncoupler of bacterial oxidative phosphorylation, were used as controls (Supplementary Fig. 8). The TMRM reagent was added to the bacterial suspensions to a final concentration of 100 nM, and the mixtures were incubated at 37 °C for 30 min. After incubation, the cells were immediately visualized by confocal laser scanning microscopy (CLSM) with excitation at 561 nm and emission detection between 576 and 683 nm.
The amounts of DNA damage in the B. subtilis strains at the different time points were evaluated via terminal deoxynucleotidyl transferase (TdT) dUTP Nick-End Labeling (TUNEL) using the In-Situ Cell Death Detection Kit with fluorescein (Roche) according to the manufacturer’s instructions. B. subtilis colonies were resuspended in PBS and processed as described above. The cells were centrifuged and resuspended in 1% paraformaldehyde in PBS and fixed at room temperature for 1 h on a rolling shaker. The cells were then washed twice in PBS and permeabilized in 0.1% Triton X-100 and 0.1% sodium citrate for 30 min at room temperature with shaking. After permeabilization, the cells were washed twice with PBS and the pellets were resuspended in 50 µl of the TUNEL reaction mixture (45 µl label solution + 5 µl enzyme solution), and the reactions were incubated for one hour at 37 °C in the dark with shaking. Finally, the cells were washed twice in PBS, counterstained with DAPI (final concentration 500 nM), mounted, and visualized by CLSM with excitation at 488 nm and emission detection between 497 and 584 nm.
Membrane fluidity was evaluated via Laurdan generalized polarization (GP)126. Colonies of the different B. subtilis strains were grown and processed as described above. The colonies were resuspended in 50 mM Tris pH 7.4 with 0.5% NaCl. Laurdan reagent (6-dodecanoyl-N,N-dimethyl-2-naphthylamine) was purchased from Sigma-Aldrich (Merck) and dissolved in N,N-dimethylformamide (DMF). Samples treated prior to staining with 2% benzyl alcohol, a substance known to increase lipid fluidity127,128, were used as positive controls (Supplementary Fig. 10). Laurdan was added to the bacterial suspensions to a final concentration of 100 µM. The cells were incubated at room temperature for 10 min, mounted, and then visualized immediately using two-photon excitation with a Spectraphysics MaiTai Pulsed Laser tuned to 720 nm (roughly equivalent to 360 nm single photon excitation), attached to a Leica SP5 microscope. Emissions between 432 and 482 nm (gel phase) and between 509 and 547 nm (liquid phase) were recorded using the internal PMT detectors.
The localization of FloT in B. subtilis cells was evaluated using a FloT-YFP translational fusion in a WT genetic background (see Supplementary Table 2 for full genotype of the strains). Colonies grown at 30 °C on MSgg solid medium were isolated at different time points and resuspended as described above. Samples were mounted and visualized immediately with excitation at 514 nm and emission recorded from 518 to 596 nm.
All images were obtained by visualizing the samples using an inverted Leica SP5 system with a 63x NA 1.4 HCX PL APO oil-immersion objective. For each experiment, the laser settings, scan speed, PMT or HyD detector gain, and pinhole aperture were kept constant for all of the acquired images.
Image analysis
Image processing was performed using Leica LAS AF (LCS Lite, Leica Microsystems) and FIJI/ImageJ104 software.
Images of live and dead bacteria from viability experiments were processed automatically, counting the number of live (green) or dead (red) bacteria in their corresponding channels. The percentage of dead cells was calculated dividing the number of dead cells by the total number of bacteria found on a field.
For processing the lipid peroxidation images, images corresponding to the reduced and oxidized channels were smoothed and a value of 3 was then subtracted from the two channels to eliminate the background. The ratio image was calculated by dividing the processed reduced channel by the oxidized channel using the FiJi image calculator tool. The ratio images were pseudo-colored using a color intensity look-up table (LUT), and intensity values of min 0 and max 50 were selected. All of the images were batch processed with a custom imageJ macro, in which the same processing options were applied to all of the acquired images. Quantification of the lipid peroxidation was performed in Imaris v7.4 (Bitplane) by quantifying the pixel intensity of the ratio images with the Imaris “spots” tool.
The Laurdan GP acquisitions were processed similarly. Images corresponding to the gel phase channel and the liquid phase channel were smoothed and a value of 10 was subtracted to eliminate the background. The Laurdan GP image was then calculated by applying the following formula (see equation 2):
$${mathrm{Laurdan}},{mathrm{GP}} = frac{{left( {{mathrm{gel}},{mathrm{phase}},{mathrm{channel}} – {mathrm{liquid}},{mathrm{phase}},{mathrm{channel}}} right)}}{{left( {{mathrm{gel}},{mathrm{phase}},{mathrm{channel}} + {mathrm{liquid}},{mathrm{phase}},{mathrm{channel}}} right)}}$$
(2)
The calculation was performed step by step using the FiJi image calculator tool. Pixels with high Laurdan GP values, typically caused by residual background noise, were eliminated with the “Remove outliers” option using a radius of 4 and a threshold of 5. Finally, the Laurdan GP images were pseudo-colored using a color intensity LUT, and intensity values of min 0 and max 1.5 were selected. This processing was applied to all of the acquisitions for this experiment. To quantify the Laurdan GP, bright field images were used for thresholding and counting to create counts masks that were applied to the Laurdan GP images to measure the mean Laurdan GP value for each bacterium.
TUNEL images were analyzed by subtracting a value of 10 in the TUNEL channel to eliminate the background. The DAPI channel was then used for thresholding and counting as described above to quantify the TUNEL signal. The same parameters were used to batch process and quantify all of the images.
To quantify the membrane potential, the TMRM assay images were analyzed as described above using the bright field channel of each image for thresholding and counting to calculate the mean fluorescence intensity in each bacterium. Endospores, which exhibited a bright fluorescent signal upon TMRM staining, were excluded from the analysis. This processing was applied to all of the acquisitions for this experiment.
To quantify the fluorescence of the bacteria expressing the floT-yfp construct, images were analyzed as described above using the bright field channel of each image for thresholding and counting to calculate the mean fluorescence intensity in each bacterium.
Statistical analysis
All of the data are representative of at least three independent experiments with at least three technical replicates. The results are expressed as the mean ± standard error of the mean (SEM). Statistical significance was assessed by performing the appropriate tests (see the figure legends). All analyses were performed using GraphPad Prism version 6. p values < 0.05 were considered significant. Asterisks indicate the level of statistical significance: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Source: Ecology - nature.com
