Reconstruction of genomes from deep-sea hydrothermal plumes
Hydrothermal vent plume and background deep-sea samples were acquired during the following cruises: R/V New Horizon to Guaymas Basin (July 2004), R/V Atlantis to Mid-Cayman Rise (Jan 2012 and Jun 2013) for Cayman Deep (Piccard) and Shallow (Von Damm) plume and background seawater samples, and R/V Thomas G Thompson to the Lau Basin (May–Jul 2009) for both plume and background seawater samples. Details of sample collection, preservation, and DNA/RNA extraction and processing are described in detail elsewhere [10, 33, 34].
In this study, we reconstructed genomes from publicly available shotgun metagenomic sequencing datasets from 19 deep-sea hydrothermal plume and surrounding background seawater samples from Guaymas Basin (Guaymas), Mid-Cayman Rise (Cayman) and Lau Basin (Lau) (Fig. 1). Additionally, we analyzed the 14 metatranscriptomic datasets that were paired with metagenomics samples from Guaymas and Cayman (Supplementary Table S1). Following quality-control, filtered reads were used to assemble scaffolds de novo according to the location of metagenomic samples. Metagenomic binning resulted in 250 metagenome-assembled genomes (MAGs) which have genome completeness >50% and genome contamination <10% in accordance with previously suggested Minimum Information about a Metagenome-Assembled Genome (MIMAG) standards [68].
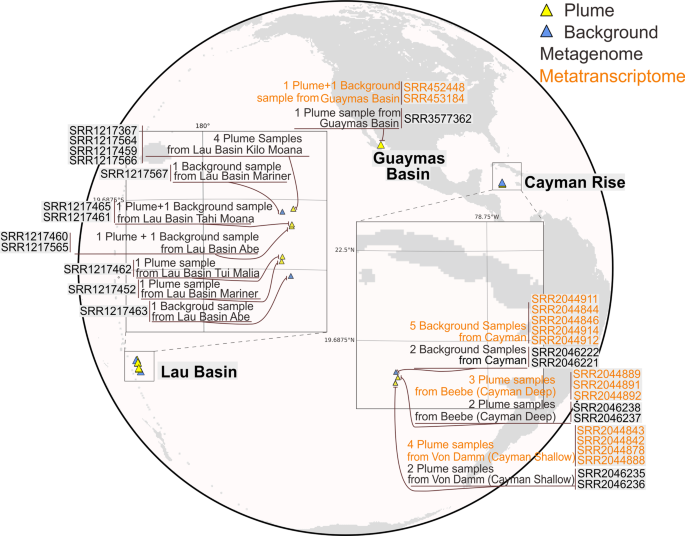
The retrieved metagenomic datasets include one from Guaymas Basin, twelve from Lau Basin, and six from Mid-Cayman Rise. Detailed sample, metagenome, and metatranscriptome information is labeled.
Phylogeny and identification of proteobacterial lineages
To identify the taxonomy of the reconstructed genomes, we used a comprehensive phylogenetic approach. First, we constructed a phylogenetic tree using a set of concatenated 16 ribosomal proteins (RP16 tree). The reconstructed phylogenetic tree revealed that bacteria comprised 219 of the 250 genomes, with 92 of them from the group Proteobacteria. We then conducted detailed taxonomic curation of all reconstructed genomes by comparison with specific databases and phylogenetic trees, namely NCBI, GTDB, and RP16. A companion 16S rRNA gene phylogenetic tree (using genes retrieved from MAGs) also has the congruent phylogeny (Supplementary Fig. S1). Of the 92 Proteobacteria genomes, we determined 51 to be phylogenetically novel (Supplementary Table S2); all lack a defined taxonomy at the scale of family, order, and/or phylum, coupled with a lack of understanding of their metabolism and ecology.
Based on the RP16 tree, we classified and defined nine proteobacterial lineages at different levels, including two phyla, three classes and four families (Fig. 2 and Supplementary Figs. S2, S3). We propose the names Marenostrumaceae for UBA2165 (since the UBA2165 type strain was first reconstructed from the Mediterranean Sea [44]; known as “Mare Nostrum” in Latin); Hyrcanianaceae for group Casp-alpha2 (since the Casp-alpha2 type strain was first reconstructed from the Caspian Sea [44]; known as the “Hyrcanian Ocean” in ancient Greek), Taraoceanobacteraceae for UBA11654, which was first reconstructed from Tara Ocean metagenome datasets [69]; Riflewellaceae for UBA4486 which was first described from terrestrial aquifer wells at Rifle, Colorado, USA [49]; Marinioligotrophales for the formerly OMG bacteria, the Oligotrophic Marine Gammaproteobacteria [70]; Planktothermales for UBA7887, which are reconstructed from hydrothermal plume environments [44]; and Kappaproteobacteria for the former LS-SOB group [44], which are ubiquitous in coastal systems and the ocean water column. This classification was also supported by the taxonomic tree associated with the GTDB taxonomy database [53]. Finally, Lambdaproteobacteria were recently described and characterized from a groundwater ecosystem [49], while this is the first study to retrieve and study genomes recovered from deep-sea hydrothermal plumes or any marine environment for this newly-discovered phylum. Most of the Proteobacteria genomes from this study have metabolic capacities associated with aerobic respiration, sulfur cycling, and CO2 fixation (Fig. 2 and Supplementary Table S3).
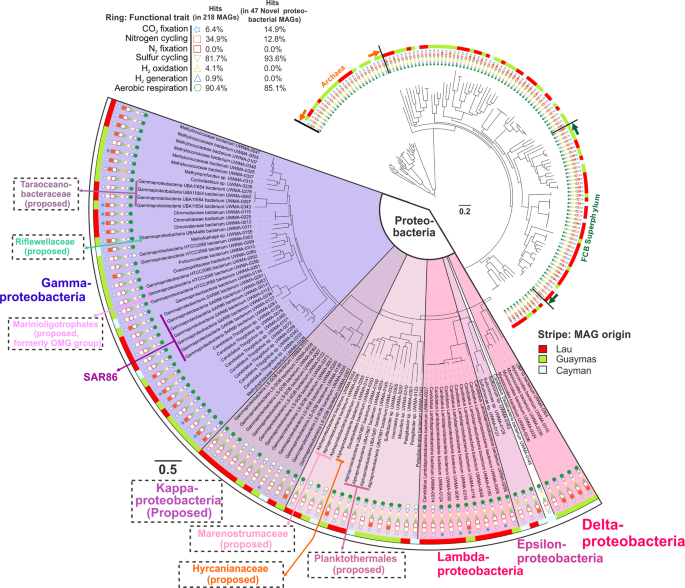
Functional traits associated with carbon, nitrogen, sulfur, hydrogen cycling, and oxygen respiration are shown. Filled and unfilled circles denote the presence/absence of function traits within a genome. This tree was visualized using iTOL [96] (https://itol.embl.de/). This figure does not necessarily reflect the branching order of all representative Proteobacteria groups.
Distribution and abundance
Novel Proteobacteria genomes identified by us constituted 23% of all reconstructed genomes from the hydrothermal environments studied here. Yet, analyses of genome coverage indicated that they comprised 36% of the microbial community, which suggests they are relatively more abundant than others (Supplementary Table S4). To determine if this abundance of Proteobacteria also translated to a higher activity in hydrothermal plumes, we compared gene expression of these organisms with members of other dominant marine phyla such as Chloroflexi (SAR202) and Bacteroidetes (Supplementary Table S4). All comparisons of metatranscriptomic mapping were conducted at the resolution of microbial groups (not individual MAGs) between novel Proteobacteria and other microbial groups. The proportion of gene expression by novel Proteobacteria was higher than that of other phyla, suggesting that novel Proteobacteria were relatively more active within community.
We then assessed the global distribution of these nine Proteobacteria lineages by examining their presence in metagenomes from the IMG/M DOE database [47]. While most of these Proteobacteria lineages were widely distributed, specifically Kappaproteobacteria, SAR86, and Marinioligotrophales were especially abundant and distributed worldwide in oceanic and coastal environments (Fig. 3). In addition, these groups were also observed in other environments outside of marine systems such as in association with a host (symbiosis), terrestrial environments, and engineered systems. Members of these Proteobacteria lineages that are abundant and active in deep-sea hydrothermal systems, are well adapted to the ambient environments and can have important ecological impacts. Finally, we used this 16S rRNA-based community survey to identify Proteobacteria lineages undergoing genome diversification and conducted comparative genomics to delineate their adaptation to specific environments.
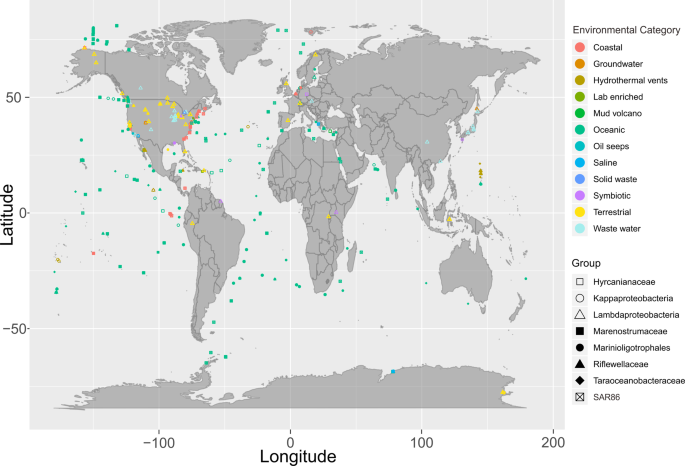
Environmental categories were parsed and summarized from “ecosystem type” information from all investigated metagenomes.
Central metabolism and respiration
We used reconstructed MAGs from this study and publicly available genomes with completeness over 80% to reflect the general metabolic and functional capacities of novel Proteobacteria (Supplementary Tables S5, S6). In addition, the metatranscriptomic datasets from Guaymas and Cayman enabled investigation of metabolic gene expression patterns (Figs. 4, 5 and Supplementary Table S6).
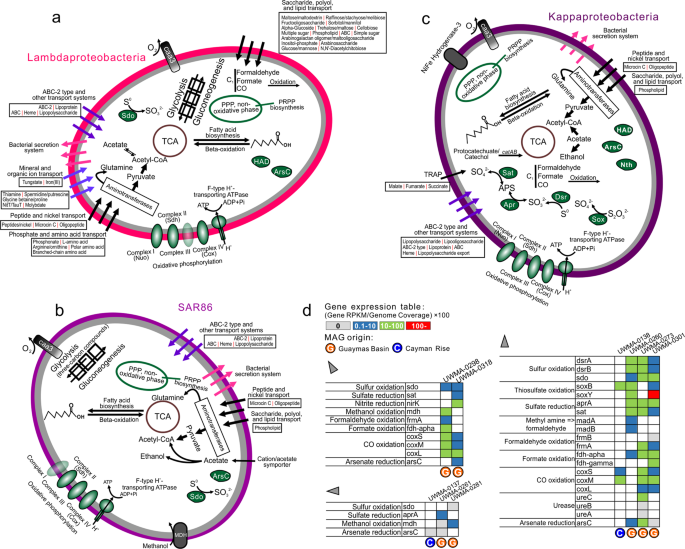
a–c Organismal profiles of metabolic capacity for Lambdaproteobacteria, Kappaproteobacteria, and SAR86, respectively. Items in gray indicate traits that are not present in over 50% of sampled genomes. d Tables indicating the normalized gene expression level of important pathways associated with energy metabolism in novel Proteobacteria genomes.
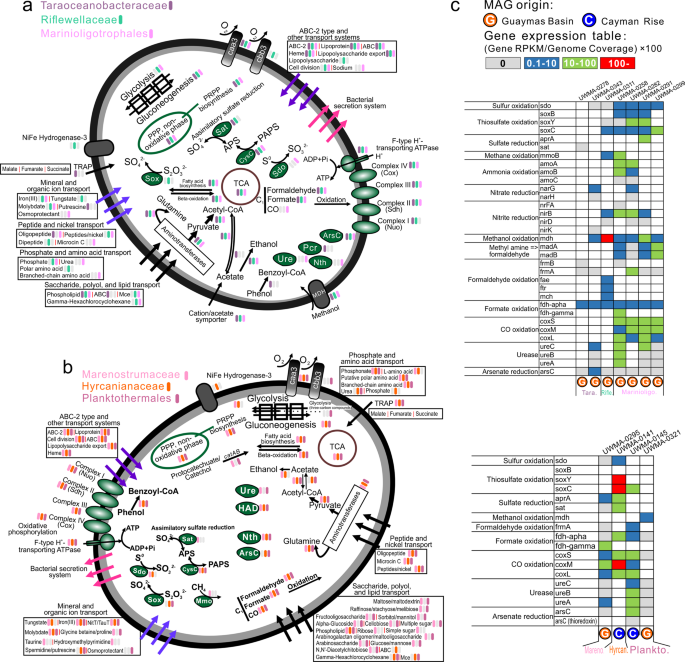
a Organismal profiles of metabolic capacities for Taraoceanobacteraceae, Riflewellaceae, Marinioligotrophales, b Organismal profiles of metabolic capacities for Marenostrumaceae, Hyrcanianaceae, Planktothermales. Items in gray indicate traits that are not present in over 50% of sampled genomes. c Tables indicating the normalized gene expression level of important pathways associated with energy metabolism in novel Proteobacteria genomes.
All nine proteobacterial lineages contained genes for central carbon metabolism pathways for biosynthesis and energy transfer: citric acid cycle (TCA cycle), glycolysis (some could only metabolize three-carbon compounds) and gluconeogenesis, peptide and amino acid utilization, pentose phosphate (PPP) pathway and PRPP biosynthesis for the generation of some nucleotide and amino acid precursors, and fatty acid biosynthesis and beta-oxidation (Figs. 4, 5 and Supplementary Table S7). These proteobacterial lineages contain either caa3/cbb3 type cytochrome c oxidases for aerobic respiration and nearly a complete set of complexes of oxidative phosphorylation for ATP generation, suggesting that fermentation and respiration can both take place.
Organic carbon metabolism
We investigated organic carbon metabolism in novel Proteobacteria by identifying the presence of transporters, secretion systems, CAZymes [71], and peptidases in the genomes. Functional predictions of cell membrane transport and secretion systems indicate that Lambdaproteobacteria, Marenostrumaceae, and Planktothermales have more transporters involved with monosaccharides, polysaccharides, polyols, and lipids which may be used as organic nutrients. Moreover, Kappaproteobacteria, Taraoceanobacteraceae, Marenostrumaceae, Hyrcanianaceae, and Planktothermales contain tripartite ATP-independent periplasmic transporters for transporting C4-dicarboxylates [72] while SAR86, Taraoceanobacteraceae, Riflewellaceae, and Marinioligotrophales contain cation/acetate symporters for direct acetate incorporation (Figs. 4, 5). Our analyses indicate that many of these organisms are capable of fermentation from acetate to ethanol and have wide-ranging capacities for the oxidation of C1 compounds such as formaldehyde, formate, and carbon monoxide. Meanwhile, Kappaproteobacteria, Taraoceanobacteraceae, Marenostrumaceae, Hyrcanianaceae, and Planktothermales can also degrade and utilize aromatic compounds, such as phenol and protocatechuate/catechol (Figs. 4, 5). Lambdaproteobacteria, Kappaproteobacteria, Marinioligotrophales, Marenostrumaceae, Hyrcanianaceae and Planktothermales contain organisms that can actively oxidize C1 compounds (only formaldehyde, formate, and carbon monoxide) from hydrothermal environments. Lambdaproteobacteria, SAR86, Taraoceanobacteraceae, Riflewellaceae, Marinioligotrophales and Planktothermales demonstrated high expression levels of methanol oxidation encoding genes. The highest expression level was observed in Riflewellaceae organisms (Figs. 4, 5). Genomes from Kappaproteobacteria and Marinioligotrophales encoded highly active genes for the utilization of methyl amines. These results indicate that these Proteobacteria are well adapted to hydrothermal ecosystems, and their metabolic activities are connected to the transformation of organic compounds of hydrothermal origin which are entrained in the plume, such as C1 (formate, formaldehyde, carbon monoxide) [13,14,15,16] and methylated compounds (methanol and methylamine) [17, 18].
To study the potential of novel Proteobacteria to breakdown carbohydrates and proteins, we screened all genomes for the presence of CAZymes and peptidases. Patterns of normalized CAZyme and peptidase gene coverage demonstrate that Lambdaproteobacteria, Kappaproteobacteria, and Marinioligotrophales are involved in carbohydrate and protein scavenging (Figs. 6, 7). Gene expression profiles suggest that Lambdaproteobacteria, Kappaproteobacteria, and Marinioligotrophales CAZymes and peptidases are highly active in Guaymas and Cayman (Supplementary Tables S9, S10). GH109 (α-N-acetylgalactosaminidase) for degradation of amino sugars, GH23 (lysozyme) for degradation of peptidoglycans and PL22 (oligogalacturonide lyase) for degradation of oligogalacturonides (a product of pectin degradation) are widely distributed in the Proteobacteria. In contrast, other families have limited distributions in specific lineages. For example, GH13 (α-amylase) for degradation of starch and pullulans, GH38 (α-mannosidase) and GH76 (ß-mannosidase) for degradation of mannooligosaccharides, and GH74 (ß-1,4-endoglucanase, endoglucanase) for degradation of cellulose and xyloglucan are primarily distributed in Lambdaproteobacteria and highly expressed. The other five proteobacterial lineages (SAR86, Taraoceanobacteraceae, Riflewellaceae, Hyrcanianaceae, and Planktothermales) exhibited a limited distribution of CAZymes and peptidases suggesting little to no involvement in carbohydrate and protein breakdown. Overall, this indicates that novel Proteobacteria participate in carbon and energy cycling in hydrothermal environments with differing metabolic strategies, which are mainly reflected in their divergent organotrophic capacities.
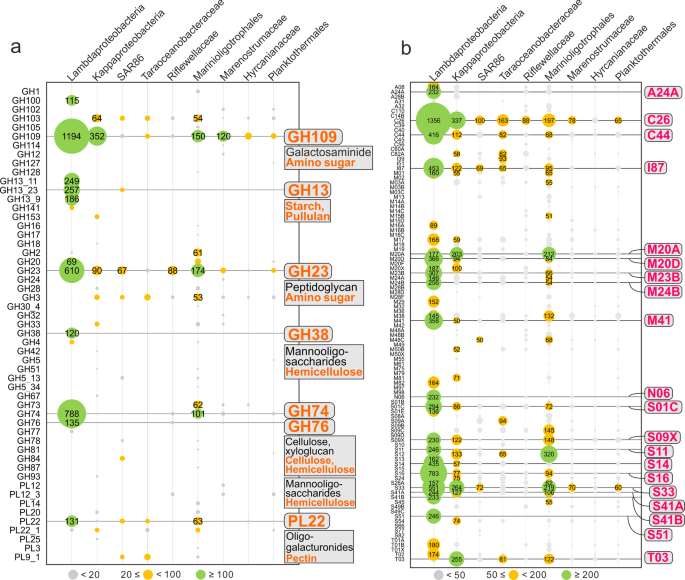
a Glycoside Hydrolase (GH) and Polysaccharide Lyase (PL) gene coverage was calculated by multiplying the identified number of CAZymes with normalized genome coverage. b Peptidase (also includes peptidase inhibitors) gene coverage was calculated by multiplying the identified number of peptidases with normalized genome coverage.
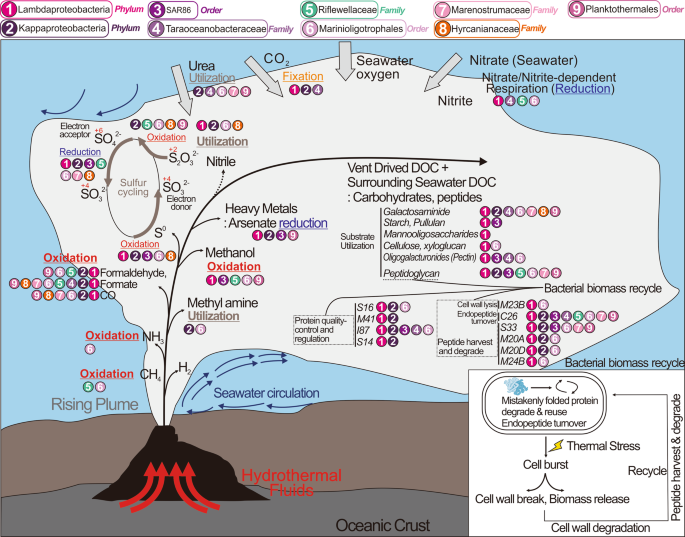
Ecological roles described here drive genome diversification. Hydrothermal plumes are shown to be distinct from the deep ocean water column. This distinction arises from temperature and density gradients of fluid masses. Only traits associated with biogeochemical cycling and energy metabolism are shown.
Lambdaproteobacteria genomes encoded the highest abundance of peptidases (Figs. 6, 7). The most abundant and actively expressed peptidases were related to protein quality control and regulation, e.g., S16 (Lon-A peptidase), an unfolded protein degrader, M41 (FtsH peptidase), and I87 (FtsH inhibitor), which inhibit FtsH and modulate the degradation of mistranslation products that disrupt membranes, and S14 (Clp peptidase), which regulates specific protein degradation (Fig. 6 and Supplementary Table S9). This could result from stress response activities that may be induced from association with high-temperature fluids [73] and serve as a protective and regulatory mechanism for cell membrane maintenance and protein transformations. Besides, there are also other abundant and active endo/extracellular peptidases in Lambdaproteobacteria which are responsible for harvesting and degrading peptides and endopeptide turnover, e.g., C26 (gamma-glutamyl hydrolase) for the turnover of folyl poly-gamma-glutamates, S33 (prolyl aminopeptidase), an extracellular peptidase prone to proline-rich substrate utilization, M20A and M20D (glutamate carboxypeptidase), and M23B (ß-lytic metallopeptidase) which lyse cell walls.
Nitrogen and sulfur metabolism
Kappaproteobacteria, Taraoceanobacteraceae, Marinioligotrophales, Marenostrumaceae, and Planktothermales genomes encode for ureases that are highly expressed suggesting that urea may be a common source of nitrogen. Nitrogen cycling activities by Proteobacteria involve both the oxidative and reductive cycle of nitrogen. Taraoceanobacteraceae, Riflewellaceae, and Marinioligotrophales encode genes associated with ammonia oxidation (amoABC) and nitrite/nitrate reduction (Figs. 4, 5). No single organism possessed the entire complement of genes for denitrification, significantly no genes were observed for nitric and nitrous oxide reduction in any genomes. Only genes for the membrane-bound Nar proteins for nitrate reduction were observed, no periplasmic Nap genes for nitrate reduction were observed in any genomes. Genes for nitrite reduction included the copper-containing nirK (for reduction to nitric oxide) and nirBD and nrfA for dissimilatory reduction of nitrite to ammonia. Many of the Proteobacteria were associated with sulfur cycling comprising the oxidative cycle of sulfur. All proteobacterial genomes lacked genes for the oxidation of hydrogen sulfide to sulfur. However, many of them encode genes for sulfur dioxygenases and reverse-dissimilatory sulfite reductases for sulfur oxidation. In particular, Kappaproteobacteria genomes encode highly expressed genes for complete oxidation of sulfur to sulfate including the dsrAB for the oxidation of elemental sulfur to sulfite and aprAB and sat for the oxidation of sulfite to sulfate (Supplementary Figs. S4, S5, and Supplementary Table S10). Kappaproteobacteria, Riflewellaceae, Hyrcanianaceae, and Planktothermales genomes possessed the Sox enzyme complex for the utilization of thiosulfate. We investigated the presence of the soxCD genes in all genomes for complete oxidation of thiosulfate in lieu of disproportionation. Amongst these, Kappaproteobacteria genomes lacked soxCD suggesting that they can only disproportionate thiosulfate to elemental sulfur and sulfate while Riflewellaceae, Hyrcanianaceae, and Planktothermales can undertake complete oxidation of thiosulfate to sulfate. Kappaproteobacteria and Hyrcanianaceae genomes exhibited high levels of sox gene expression suggesting active utilization of thiosulfate in hydrothermal plumes (Figs. 4, 5 and Supplementary Table S10). Overall, these results suggest that these novel Proteobacteria actively oxidize and cycle various nitrogen and sulfur species as nutrient and energy sources [11].
Metabolism of iron and arsenic
Genes encoding for mineral transport enzymes for Fe (III) could be found in the Lambdaproteobacteria, Riflewellaceae, Marinioligotrophales, Marenostrumaceae, Hyrcanianaceae and Planktothermales genomes, suggesting that these Proteobacteria likely participate in the acquisition of Fe from precipitating minerals in hydrothermal plumes. Microorganisms store iron within the cell by reducing Fe (III) to Fe (II), and incorporate iron into a variety of organic compounds by forming C–Fe or S–Fe bonds, such as in metalloproteins, ferredoxins, and NADH dehydrogenase which are of significant importance to cellular activities [74]. In doing so, plume Proteobacteria may serve as a part of the microbial Fe pump to scavenge and store mineral-bound Fe in biomass, to sequester Fe in the organic carbon pool after cell death, and to transfer Fe widely as plumes disperse across the oceans [75].
Arsenic and arsenic minerals are discharged in hot, mineralized hydrothermal fluids [76], and organisms in close proximity to the rising plume need to be resistant to elevated arsenic concentrations. Nearly all proteobacterial genomes contained genes for arsenate reduction (arsC), and many arsC genes have considerably high expression in hydrothermal plumes, such as in Planktothermales at Cayman, and Taraoceanobacteraceae and Kappaproteobacteria at Guaymas. Arsenic resistance and detoxification are mediated by arsC and highly abundant arsC genes have previously been found in hydrothermal systems such as at the iron-rich hydrothermal microbial mats from Lō’ihi Seamount, Hawai’i [77].
Linking genome diversification to adaptation of functional traits
Microbial traits often evolve in close coordination with their environment. For instance, biogeographical distribution of Prochlorococcus suggests that some genome contents are associated with specific regions and environmental factors, such as low nutrients and temperature [78]. Therefore, we investigated genome diversification of individual clades of these nine proteobacterial lineages in the context of niche-specific metabolic functions. We specifically identified proteobacterial lineages from hydrothermal environments that were also observed in other broad marine and terrestrial environments and performed the comparative genomic analyses of these genomes. Amongst the lineages investigated by us, SAR86, Kappaproteobacteria, Muproteobacteria, and Lambdaproteobacteria exhibited the strongest evidence of genome diversification and its association with environmental adaptation.
SAR86
SAR86 is a globally distributed clade of heterotrophic Proteobacteria. Here, we have provided some of the first evidence elucidating the roles and importance of SAR86 in hydrothermal plumes. Our concatenated ribosomal protein-based phylogenetic tree for SAR86 indicated the presence of three distinct clades, namely the non-photic zone clade that also harbors hydrothermal plume derived genomes, the photic zone clade, and the marine subsurface clade (Fig. 8a), which are associated with the specific environments they inhabit. To understand genome diversification associated with this niche differentiation, we examined clade-specific genes and metabolic pathways in these SAR86 clades. All three clades possess a unique distribution of orthologous genes with specific functions that point to their genome diversification (Fig. 8a). The photic zone clade exclusively possesses proteorhodopsins as the light-driven proton pump for energy conservation which is important for their limited chemoorganotrophic lifestyle [79]. In spite of the importance of proteorhodopsin in these organisms, they lack the ability to synthesize retinol as a chromophore [80, 81]; in line with previous reports, we hypothesize that short-chain dehydrogenases are responsible for converting other substrates (e.g., retinal or β-carotene) to retinol [80]. The aquatic β-propeller phytase (BPP) enzymes are found in marine bacteria for the mineralization of phytate to recycle phosphorus [82]. We observed the presence of BPP, phosphate-starvation-inducible protein, alkaline phosphatase [83] and phospholipase only in the photic zone clade suggesting that they are more subjected to a phosphorus-limited environment and have evolved to gain a set of response mechanisms against phosphorus-starvation. Finally, the exclusive distribution of DNA photolyases (PhrB/-like) in the photic zone also indicates that SAR86 in this environment have evolved to repair DNA damage caused by exposure to ultraviolet light [84] (Fig. 8a).
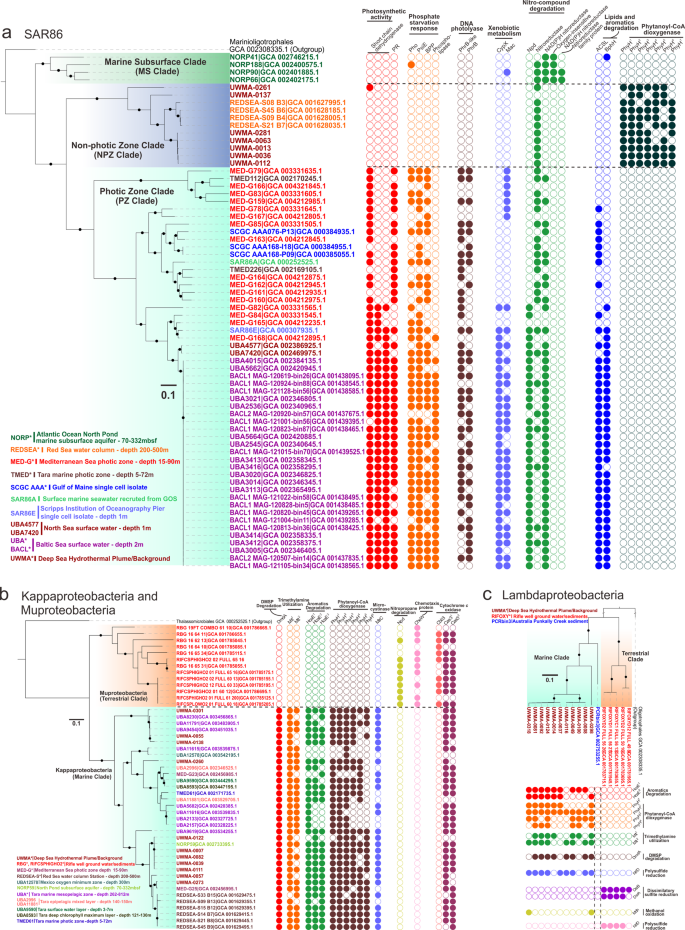
Phylogenetic trees were reconstructed based on concatenated 16 RPs, and branches with bootstrap values [ultrafast bootstrap (UFBoot) support values] over 90% by IQ-TREE were labeled with black dots. The tree scale is provided in each sub-panel with 0.1 amino acid substitutions per site, accordingly. Each circle on the panel right to the tree indicates an ortholog group (OG) with a specific function. Filled circles represent the presence and unfilled circles represent the absence of specific traits.
Genes unique to the non-photic zone clade (that includes genomes from hydrothermal environments) of SAR86 include phytanoyl-CoA dioxygenases (PhyH) associated with the breakdown of chlorophyll. The copy number of phyH in this clade ranged from 4 to 7. PhyH could hydroxylate the methyl-branched chain of phytanoyl-CoA and is required in the alpha-oxidation pathway of fatty acid metabolism to move the methyl-group from beta position to the alpha position [85]. Together with the metabolism of alcohol and aldehyde dehydrogenases and acyl-CoA synthetase, it is feasible for bacteria to transform phytol (a long-chain alcohol constituent of chlorophyll) to phytanoyl-CoA and pass the products downstream towards beta-oxidation and energy utilization [85, 86]. The exclusive distribution of PhyH in the non-photic zone clade implicates that this specific group could potentially scavenge the degradation products of chlorophyll for carbon and energy demand, which could possibly be of phytoplankton-origin from the upper ocean layers (e.g., deep chlorophyll maximum) [87]. Macrolide transporter and cytochrome P450 are potentially responsible for the degradation of xenobiotics (non-naturally produced compounds toxic to microbes, e.g., therapeutic drugs, antibiotics) [80], and the genes encoding for them are only found in photic zone genomes (Fig. 8a). It also indicates that photic zone genomes possess nitronate monooxygenases and nitroreductases for degrading nitronate and nitro-containing compounds (e.g., nitroaromatics and nitroheterocyclic compounds) (Fig. 8a). Furthermore, photic zone genomes possess additional capacities to degrade long-chain fatty acids and aromatics (Fig. 8a). This potentially indicates that xenobiotic compounds, nitro-compounds, lipids, and aromatics are universal in the surface oceans and surface microbes are adapted for biological defense and substrate utilization. SAR86 inhabiting the marine subsurface are also capable of degrading nitro-compounds, as their genomes contain different types of nitroreductases, such as NAD(P)H dependent, oxygen-insensitive nitroreductases, and other nitroreductase family proteins (Fig. 8a).
Kappaproteobacteria and Muproteobacteria
Our genome diversification analysis suggests that two lineages, Kappaproteobacteria and Muproteobacteria evolved to uniquely inhabit specific environments, namely terrestrial (Muproteobacteria) and marine (Kappaproteobacteria). The two lineages are closely related, representing monophyletic deep-branching clades that likely share a common ancestor (Supplementary Fig. S2). Kappaproteobacteria genomes encode important functional traits for utilizing elemental and energy sources in the ocean (Fig. 8b). Dimethyl sulfoniopropionate (DMSP) is a widely distributed marine algal osmolyte and is well known as a significant source of carbon and sulfur for bacterioplankton [88], while trimethylamine (TMA) is part of the oceanic organic nitrogen pool and produced by reduction of many marine osmolytes such as glycine betaine, trimethylamine oxide (TMAO), and choline [89]. Kappaproteobacteria genomes encode enzymes to utilize all these above-mentioned compounds. Similar to SAR86, Kappaproteobacteria can also potentially utilize aromatics and chlorophyll degradation products in the ocean. Furthermore, microcystin, as a group of toxins produced by cyanobacteria, could be found not only in freshwater but also marine environments [90]. Kappaproteobacteria contain genes encoding for microcystinase which could degrade and detoxify microcystin that is generated by marine cyanobacteria [91]. These results suggest that Kappaproteobacteria are well adapted to life in the marine water column.
In contrast, Muproteobacteria genomes were primarily sourced from terrestrial ground water and sediments and were absent in marine environments [49]. Microbes encoding chemotaxis proteins (CheW) are better adapted to sense and respond to the chemical gradients beneficial to their survival in porous underground environments. Nearly all Muproteobacteria genomes we examined possessed CheW. Muproteobacteria also encode genes to utilize nitropropane (a potential industrial hazardous compound) from terrestrial environments (Fig. 8c). Despite their existence in terrestrial environments with limited oxygen, Muproteobacteria possess cytochrome c oxidases and other complexes for aerobic respiration (Supplementary Table S11). Muproteobacteria genomes contain both the caa3 type (also referred to as A-type) and cbb3 type (also referred to as C-type) cytochrome c oxidases, while Kappaproteobacteria only contain the caa3 type. The cbb3 type cytochrome c oxidase has a higher oxygen binding affinity in comparison to caa3 and helps microorganisms to respire under low-oxygen conditions [92]. These results provide a genome-based understanding of Muproteobacteria as a lineage of organisms that are highly adapted to survive and proliferate in the terrestrial subsurface.
Lambdaproteobacteria
Similar to Kappaproteobacteria and Muproteobacteria, the marine clade of Lambdaproteobacteria contains genes for degradation of aromatics, chlorophyll degradation products, DMSP, and TMA (Fig. 8). The functional trait of methanol oxidation is essential for utilizing methanol entrained in the hydrothermal plume as indicated (Fig. 8). Less well-known is their role in terrestrial environments and the metabolic differences between the marine and terrestrial clades. Both the terrestrial and marine clades of Lambdaproteobacteria contain genes for the reduction of polysulfide, however, the enzymes belong to different protein families, which suggests that they might operate differently in corresponding environments to deal with polysulfide reduction, e.g., with different substrate binding affinities, different enzymatic activities and/or adaptation to salinity (Fig. 8). Finally, only organisms from the terrestrial clade of Lambdaproteobacteria possess the dissimilatory sulfite reductase pathway to reduce sulfite to hydrogen sulfide [93].
Source: Ecology - nature.com

