Sampling sites
Thirteen sites in streams of 3 cantons of Switzerland and 7 sites along the shores of Lake Geneva were selected to cover a known gradient of anthropogenic pressures, in order to compare the morphological and metabarcoding approaches for determining the oligochaete indices (Supplementary Table S1). The 13 stream sites comprised sources and sites in industrial, urban and agricultural areas. The 7 sites in Lake Geneva were selected based on a previous study on the physicochemical quality of sediments of this lake22 with sediments containing a range of metal concentrations.
Sampling and sieving of samples for morphological and genetic analyses
Sediment samples (3 L) were collected and sieved according to IOBS and IOBL protocols2 using a Surber type net (0.2 mm mesh size) for stream sites and an Ekman type grab sampler for lake sites. The top 10 cm of sediments were collected in both stream and lake sites. The water depth where the samples were collected was 30–50 cm for stream sites and 20–70 m for lake sites. At each site (streams and lake), 3 subsamples (one sample every 10–20 meters) were collected, combined and fixed with 10% neutral buffered formalin (ThermoFisher Scientific, Ecublens, Switzerland) adjusted to a final formaldehyde concentration of 4%. Formalin optimally fixes oligochaete specimens, and a study showed that fixation and storage of oligochaete specimens in 4% neutral buffered formalin for up to 30 days was suitable for subsequent genetic analyses23. Sediment samples were stored at 4 °C for 1 to 5 days before sieving, then sieved through a column of sieves with 5 mm and 0.5 mm mesh size. The material retained on the 0.5 mm sieve was transferred to a plastic box and preserved in absolute ethanol at −20 °C. The big oligochaete specimens retained in the 5 mm sieve were incorporated in the plastic box.
Morphological analysis
Morphological analyses of oligochaete samples followed IOBS and IOBL guidelines2. The preserved specimens were transferred to a subsampling square box (5 × 5 cells), and the contents of randomly selected cells were transferred into a Petri dish and examined under a stereomicroscope until 100 specimens were collected. In two samples from the sources of streams, oligochaete densities were very low and only 28 (source Boiron) and 43 specimens (L’isle) per site were obtained for morphological analysis. In these samples, the identification of low numbers of specimens had no impact on the results of ecological diagnoses as all specimens belonged to sensitive species indicating good sediment quality. Sorted specimens were then mounted on slides in a coating solution composed of lactic acid, glycerol and polyvinylic alcohol. Oligochaete specimens were identified to the lowest practical level (species if possible) using a compound microscope.
Genetic analysis
Sorting of specimens
As for morphological analysis, sieved material preserved in absolute ethanol at −20 °C was transferred into a subsampling square box (5 × 5 cells). The contents of randomly selected cells were examined under a stereomicroscope until 33 (9 sites) or 66 specimens (11 sites) were collected (Supplementary Table S1). We assumed that the identification of 33 specimens (corresponding to about 1/3 of the number of morphologically identified specimens) to the species level per site could be enough for assessing the biological quality of sediments. For the 11 sites where 66 specimens were collected, we sorted out two sets of 33 specimens to examine the differences obtained in species diversity and abundance by analysing 33 or 66 specimens, and to determine if the analysis of only 33 specimens was sufficient to establish correct ecological diagnoses. The posterior region of each specimen was cut off transversally and transferred to a 1 ml tube containing 0.03 ml absolute ethanol (1 tube per specimen). The anterior part of some specimens (12 to 24 per site) was preserved in absolute ethanol for further morphological identification, in case some of the preserved specimens corresponded to new lineages (=species), i.e. were unassigned using our Swiss COI reference database5 or GenBank public database (NCBI). Specimen samples for genetic analysis were stored at −20 °C until DNA extraction. Before DNA extraction, samples were dried for 24 hours at room temperature. The workflow from fixation of specimens to calculation of genetic indices is shown in Fig. 1.
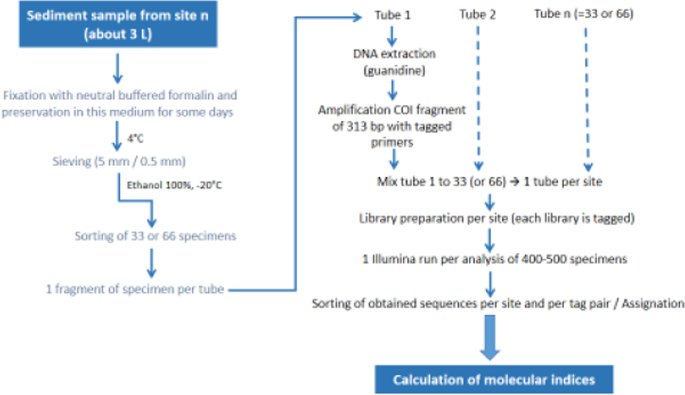
Workflow of the different steps of the high-throughput DNA barcoding analysis from fixation of oligochaete specimens to calculation of the indices.
DNA extraction, PCR amplification, library preparation and Illumina sequencing
Total genomic DNA was extracted from tissue samples using the guanidine thiocyanate method described by Tkach and Pawlowski (1999)24. The primers specific to metazoans “mlCOIintF” and “jgHCO2198”25 were used to amplify a COI fragment (313 base pairs) from each DNA extract. PCR amplifications were performed in a total volume of 50 μl containing 0.5 μl of Taq polymerase 5U/μl (Roche, Basel, Switzerland), 5 μl of the PCR buffer (10x concentrated) with MgCl2 (Roche), 1.25 μl of each primer (10 μM each), 1 μl of a mix containing 10 mM of each dNTP (Roche) and 2.5 μl of DNA template. The PCR comprised an initial denaturation step at 95 °C for 5 min, followed by 35 cycles of denaturation at 95 °C for 40 s, annealing at 44 °C for 45 s and elongation at 72 °C for 1 min and a final elongation step at 72 °C for 8 min.
The metazoan primers were tagged by bearing 8 nucleotides attached at each primer’s 5′ extremity. A unique combination of tagged primers was used for each specimen in order to multiplex all specimens in a unique sequencing library26. PCR products of each specimen were quantified with capillary electrophoresis using QIAxcel instrument (Qiagen, Hilden, Germany). Equimolar concentrations of PCR products were pooled into a single tube (one tube per site) and purified using High Pure PCR Product Purification kit (Roche Diagnostics). Then, 700 ng of purified PCR products was appended with Illumina PE adapter sequences in order to obtain one functional sequencing library per PCR sample (or site). This was performed using the TruSeq DNA PCR-Free Library Preparation Kit (Illumina) following the kit instructions to include a unique index as a label for each library. The libraries were quantified by qPCR using the KAPA Library Quantification Kit (Roche). Finally, libraries were sequenced on a MiSeq instrument using paired-end sequencing for 500 cycles with nano kit v2.
The raw sequences are accessible in the Short Read Archive under the BioProject number PRJNA563268. The COI sequences of 313 bp corresponding to new lineages for our local reference COI database (one sequence per lineage) obtained as part of this study are provided in Supplementary File S1. In the near future, we will Sanger sequence a large segment of COI (658 bp) using universal primers27 of the specimens corresponding to these lineages. We will deposit these sequences (658 bp) in the European Nucleotide Archive as part of a future publication on the update of our COI reference database.
Analysis of sequences
Bioinformatic analyses were performed using an in-house pipeline (SLIM28). Raw fastq reads were quality-filtered by removing any sequence with a mean quality score of 30 and removing all sequences with ambiguous bases or any mismatch in the tagged primer. Paired-end reads were then assembled using simple bayesian algorithm implemented in PANDAseq29. Chimera removing and the OTUs clustering at 97% were performed using VSEARCH30.
To assign the sequence corresponding to each tagged specimen to a specific lineage (or species), we considered that the sequences diverging by less than 10% (in COI) belonged to the same species, except for some species within the genera Nais and Uncinais (8%)5. The obtained sequences were first taxonomically assigned based on our local COI reference database5 using VSEARCH algorithms30. Then, each unassigned sequence using our local reference database was taxonomically assigned based on GenBank database using BLAST (http://www.ncbi.nlm.nih.gov/BLAST/Blast.cgi). Sequences that could not be assigned using these databases were identified either through morphological analysis of corresponding anterior part or by building barcode trees. Specimens taxonomically assigned using such trees were identified to the family or subfamily level. To construct these trees, the neighbour-joining method as implemented in Seaview v.4.4.0 was applied31, with 1,000 bootstrap replicates. The genetic distances between our sequences and GenBank’s sequences and between the sequences taxonomically assigned by building barcode trees were calculated using the K2P model in MEGA 5.132.
Oligochaete indices
To analyse the genetic data, we applied the same index calculations (for streams and lake) as for the morphological analysis:
For assessing the biological quality of fine/sandy sediments of streams we applied the IOBS index2,3 calculated according to the following formula:
$${rm{IOBS}}=10{{rm{ST}}}^{-1}$$
where S is the total number of taxa identified among 100 oligochaete specimens examined per sample, and T is the percentage of tubificids with or without hair setae that is dominant in the sediment sample (mature and immature worms combined). The index ranks the biological quality of sediments as follows: IOBS ≥ 6: very good, 3–5.9: good, 2–2.9: medium, 1–1.9: poor, <1: bad.
The biological quality of lake sediments was assessed using the “percentage of sensitive taxa” to pollution, as described in the IOBL guideline, which also contains a list of sensitive oligochaete taxa in lakes2,4. To this list, we added the species Spirosperma ferox, considered in lakes as sensitive by Lods-Crozet & Reymond (2005)33. The biological quality is ranked as follows: percentage >50: very good, 21–50: good, 11–20: medium, 6–10: poor, 0–5: bad. The IOBL index itself was not calculated. This index assesses the functioning of the lake sediments (ranks from low to high metabolic potential) and takes into account the total number of taxa and oligochaete density4. It is therefore not suitable for a comparison between morphological and genetic results as the two approaches would be compared only based on the total number of taxa.
Statistical analyses
Linear regressions between the IOBS index values, percentages of sensitive taxa (lake) and percentages of the families/subfamilies that were frequent in our samples (Tubificinae with hair setae, Tubificinae without hair setae, Naidinae, Pristininae, Lumbriculidae and Enchytraeidae) obtained using the morphological and genetic data were performed. For each relationship, we determined the coefficient of determination R2, the slope (a) of the linear regression line genetic (y)/morphology (x) (y = ax + b) and applied the Pearson test. These analyses were performed using the Free Statistics and Forecasting Software34. Prior to statistical analysis, a log-linearization was applied to the IOBS data.
Source: Ecology - nature.com

