Growth of A. woodii
A. woodii DSM1030 was cultivated at 30 °C under anoxic conditions in complex medium as previously described [18]. When using the pyrE deletion mutant, 50 mg/l uracil was added to the medium [12]. Unless otherwise stated 20 mM fructose or 20 mM fructose together with H2 + CO2 (80:20 [v/v]) were used as carbon source. Gaseous substrates were used at a pressure of 1.0 × 105 Pa. For growth experiments, concentrations of the used carbon sources were as follows: lactate, 80 mM; ethanol, 50 mM; formate, 100 mM. Minimal medium used for genetic manipulations was prepared as previously described [19] in which yeast extract was omitted and higher amounts of 0.2 g l−1 KH2PO4, 1.35 g l−1 NH4Cl, and 1.5 ml l−1 of selenite/tungstate solution were used and 10 µg ml−1 of D/L-pantothenate was added. For mutant selection, 1.7 µg ml−1 uracil and 1.5 mg ml−1 5-fluoroorotic acid were added.
Genetic modifications
For deletion of the hydBA genes, an uracil auxotroph pyrE deletion mutant was generated using the suicide plasmid pMTL_AW_KO1. The background of the plasmid pMTL_AW_KO1 is pMTL84151 [20] out of which the Gram+ origin of replication was partially deleted by digestion of the vector with XmnI and FspI following a blunt-end ligation. The pyrE deletion cassette was cloned into the multiple cloning site, consisting of a 393 bp upstream flanking region including the first 52 bp of pyrE and a 399 bp downstream flanking region including the last 68 bp of pyrE. Both flanking regions were amplified via PCR, joined by splice-by-overlap-PCR (SOE-PCR) and cloned into the plasmid using BamHI and EcoRI. Transformation and integration of the plasmid into the A. woodii wild type as well as recombination of the plasmid at its homologous regions toward the loss of the pyrE gene has already been described elsewhere [12]. The deletion of 456 bp of the pyrE gene was verified by DNA sequencing analysis [21]. The suicide plasmid pMTL_AW_KO2 for the in-frame deletion of hydBA genes in A. woodii was built in the pMTL_AW_KO1 background. First, the pyrE cassette from Eubacterium limosum KIST612, consisting of the pyrE gene (ELI_0961) and 66 bp of its promoter region, was placed behind the catP resistence marker cassette to be used as a counter selectable marker as described previously [12], generating pMTL_AW_KO1_pyrE_Elim. Second, both fragments of the hydBA deletion cassette, consisting of a 486 bp upstream flanking region including the first 18 bp of hydB and a 462 bp downstream flanking region including the last 15 bp of hydBA, where amplified via PCR, ligated via SOE-PCR and were cloned into pMTL_AW_KO1_pyrE_Elim by using EcoRI and XbaI, which replaced the original pyrE deletion cassette with the hydBA deletion cassette. For plasmid transformation into the pyrE mutant and further integration and recombination of the hydBA deletion cassette, the same protocol as for the pyrE gene deletion, as described above, was followed. The deletion of 3538 bp of the hydBA region was verified by DNA sequencing analysis [21].
Preparation of cell-free extracts
Cells were harvested in the late exponential growth phase and resuspended in lysis buffer, containing 25 mM Tris-HCl buffer, pH 7.8, 420 mM sucrose, 2 mM DTE, 4 µM resazurin, for a 1 h treatment with 2.8 mg ml−1 lysozyme. After washing the protoplasts in analytical buffer, containing 25 mM Tris-HCl, pH 7.6, 20 mM MgSO4, 20 % [v/v] glycerol, 2 mM DTE, 4 µM resazurine, 0.5 mM PMSF, 0.1 mg ml−1 DNAseI, protoplasts were passed twice through a french pressure cell at 110 MPa (Thermo, Needham Hights, MA, USA). The cell-free extract was separated from cell debris by centrifugation at 12,000 × g for 15 min.
Hydrogenase activity assays
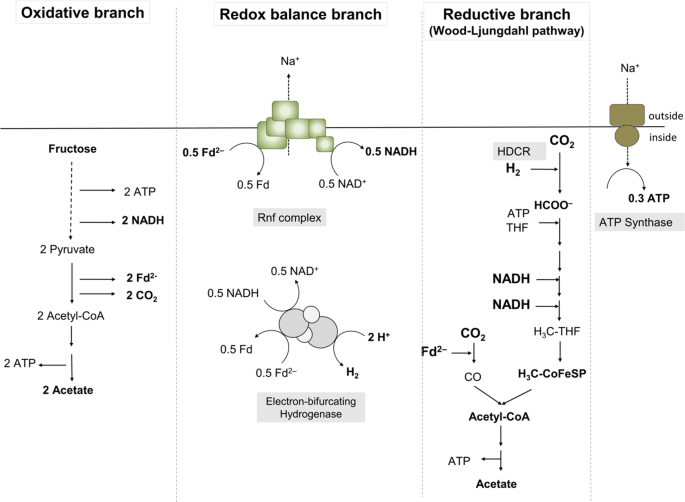
The activity of the electron-bifurcating hydrogenase was determined as described previously [22].
Western blot analysis
For detection of HydB and HydA subunits in cell-free extracs, Western blot analysis was performed as described before [23].
Stirred-tank reactor cultivations
Bioreactor cultivations were conducted in a 2 l working volume Biostat Aplus fermenter (Sartorius, Melsungen, Germany). The vessel was equipped with temperature probe, sparger, baffles, two Rushton-impeller, pH-probe (Hamilton, Bonaduz, Switzerland) and a redox potential probe (Hamilton, Bonaduz, Switzerland). The gas stream into the reactor was maintained at a constant rate by a digital mass-flow controller (Bronkhorst High-Tech, Ruurlo, Netherlands).
Analytical methods
Protein concentrations were determined by the method described by Bradford [24]. Fructose, formate and acetate concentrations were measured enzymatically (Hoffmann-La Roche, Basel, Switzerland). Hydrogen concentrations were determined by gas chromatography (GC) as described previously [17, 25]. Fermentation off-gas analysis was conducted by a Micro-GC (Inficon, Bad Ragaz, Switzerland) equipped with two measurement modules. Module one had argon as carrier gas for determination of hydrogen, oxygen and nitrogen, module two had helium as carrier gas for determination of carbon dioxide and water. Sampling time of the GC was 25 s. The analytical conditions for module one were: injector temperature, 90 °C; column pressure, 2.07 × 105 Pa; column temperature, 80 °C, column, Rt-Molsive 5 Å, 0.25 mm ×10 m with a Rt-Q-Bond, 3 m precolumn and for module two: injector temperature, 90 °C; column pressure, 1.72 × 105 Pa; column temperature, 60 °C, column, Rt-Q-Bond, 0.25 mm × 8 m. Each module was equipped with a thermal conductivity detector and the sampling rate was set to 100 Hz. Analysis time was 90 s for both modules. Off-line samples were taken in 2 ml volume. OD600 was measured in a spectrophotometer, Genesys 10 s UV-Vis (Thermo Scientific, Madison, USA) at a wavelenth of 600 nm. Samples were diluted with 0.9 % NaCl solution, if necessary. Samples were centrifuged at 16,363 × g for 10 min and the supernatant was used for analysis of acetic acid, formic acid and fructose using enzymatic assays as described above.
Source: Ecology - nature.com

