Microbial community structures
Multiple sites were sampled across a range of depths for direct comparison: one blue hole site (SYBL), two ocean sites (C3 and C4), and three lagoon sites (C1, C2, and C5; Fig. 1; Table 1). We successfully sequenced 40 PCR samples, and high-quality sequence reads were generated for further analysis. Overall, 16, 596 operational taxonomic units (OTUs) were identified across all samples, with some samples having up to 3, 595 OTUs.
Based on redundancy analysis (RDA, Fig. 2), environmental data explained 83.5% of total community variation of the Sansha Yongle Blue Hole and surrounding open sea waters at the phylum level, where RDA1 explained 60.5% of the variation and RDA2 explained 23.0% of the variation. O2 was the most important factor affecting the microbial assemblages, and significantly explained 44.8% of the total variation, followed by silicate (44.2%), temperature (38.9%), H2S (34.8%), NH4+ (31.2%), CH4 (28.2%), nitrous oxide (N2O, 22.3%), NO3− (12.8%), dissolved organic carbon (DOC, 12.4%), salinity(11.7%), POC (9.2%), and chlorophyll a (7.3%) (P < 0.05). On the basis of the cluster analysis, all samples fell into three groups at the phylum level (Fig. 3a). The first group consisted of samples located between 0 m and 30 m at C5, 0 m and 10 m at C1, 0 m and 30 m at C2, 0 m and 10 m at SYBL, 0 m and 150 m at C3, and 0 m and 75 m at C4. The samples at SYBL, C3, and C4 within this group had high levels of O2, DOC, POC, and chlorophyll a (Fig. 2). High levels of cyanobacteria were also observed within this depth range (Fig. 3b). The theoretical euphotic layer (1% of surface irradiance) was 51.2 m at SYBL and 80.8 m at C4, suggesting the samples in the first group at SYBL and C4 were located above the euphotic layer. Thus, these samples were characterized by high primary productivity and O2 enrichment via a light-driven process. The second group included samples located between 200 m and 300 m at C3, 100 m and 300 m at C4, and 30 m and 90 m at SYBL. These samples were from the depths with lower O2 concentration and higher NO3− level, when compared with the first group, implying NO3− accumulation and transformation. Samples in the third group were distributed among the anoxic bottom layer of the blue hole (100–300 m) and were characterized by high levels of H2S, NH4+, and CH4, suggesting highly reductive. The oxic-to-suboxic zone (30–90 m) in the blue hole displayed a similar microbial composition with deep waters of the surrounding open sea when compared with the anoxic bottom layer. This result is consistent with the observation that the functional capability of microbial communities at the shallow Landsort Deep of the Baltic Sea was similar to those of two deep communities: the 6 km-depth of a trench off Puerto Rico and the 1 km-depth of the Marmara Sea47. The similarities were both likely due to the stagnant conditions and hypoxia that shifted towards the surface of the water column47. Therefore, biochemical processes in deep waters might occur in shallow waters under O2 deficiency.
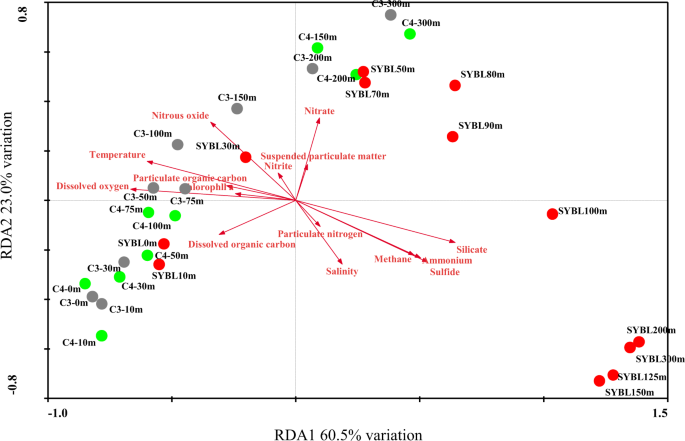
Redundancy analysis (RDA) of 70 microbial phyla from the Sansha Yongle Blue Hole and surrounding regions, based on 16S rRNA amplicon sequences.
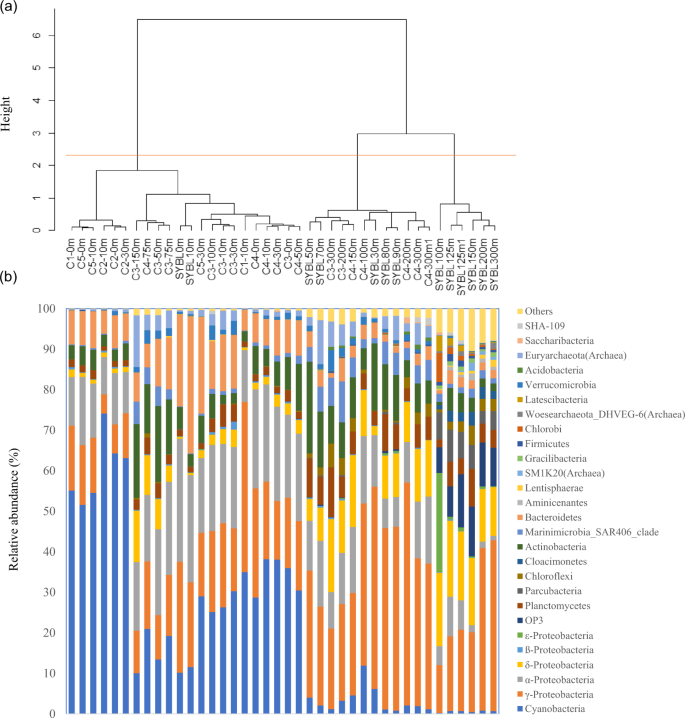
(a) Cluster analysis and (b) relative abundance of microbial phyla from the Sansha Yongle Blue Hole and surrounding regions, based on 16S rRNA amplicon sequences.
Samples were also recovered in three groups at the genus level, although some samples (C4-75 m, C3-50 m, and C3-150 m) were clustered with the deep-water open sea samples (Fig. S1). This suggested that the abundance and composition of the microbial genera shifted at the PNM (SYBL: 30 m; C3: 100–150 m; and C4: 75–100 m). Interestingly, the blue hole samples within each of the three branches formed separate sub-branches. This indicated that at high taxonomic level, microbial composition in the blue hole differed from that in the surrounding water. Microbial composition also varied throughout the water column, with distinct sub-divisions partitioned along the chemocline. Therefore, microorganisms occupied different niches in the blue hole that could be linked to different biogeochemical processes.
Microbial composition and distribution
Surface layer
Based on the 16S rRNA amplicons, Cyanobacteria (10.9%), α-Proteobacteria (28.4%), γ-Proteobacteria (24.3%), Bacteroidetes (28.1%), and Actinobacteria (4.2%) were dominant at 0 m and 10 m in the blue hole (Fig. 3b). These populations are typical in marine environments, including the oxic surface waters overlying OMZs46,48. Consistently, metagenomic sequences of Cyanobacteria (25.4%), α-Proteobacteria (26.1%), γ-Proteobacteria (28.5%), and Bacteroidetes (5.4%) were dominant at 10 m in the blue hole (Fig. S2). The relative abundance of Cyanobacteria in the surface layer was 10.2% and 11.5%, which is similar to C3-150 m and C4-100 m. The extinction coefficient of visible light in the blue hole was higher than in the open sea and this rapid attenuation of light might limit cyanobacterial growth. We detected sequences affiliated with α-Proteobacterial class Rhodobacteraceae (relative abundance, 22.9% and 20.7%). Rhodobacteraceae-affiliated sequences were also abundant in the oxic surface waters overlying OMZs, including the Saanich Inlet and the ETSP46. Many Rhodobacteraceae species are known for their close associations with algal blooms, as well with particles49,50,51, and preferentially use labile organic substrates51. Bacteroidetes are the most abundant phylum in the world ocean after Proteobacteria and Cyanobacteria. In the blue hole, Flavobacteriales sequences accounted for a majority of Bacteroidetes and were most abundant at 0 m (22.2%) and 10 m (33.3%). This is consistent with the abundance of Bacteroidetes in other coastal areas (10–30%)52. Flavobacteriales are often associated with marine snow and marine phytoplankton blooms50,53,54. These bacteria attach to phytoplankton aggregates and efficiently degrade and preferentially consume high-molecular-mass organic matter as primary carbon and energy sources51.
Intermediate layer
Between 30 m and 90 m, the blue hole exhibited a sharp oxycline: from oxic (30–70 m), to hypoxic (80 m), and then to suboxic (90 m). The prevalent 16S rRNA amplicons across this transition included those affiliated with the γ-Proteobacteria (24.4–49.9%), Actinobacteria (11.3–22.6%), α-Proteobacteria (7.3–16.3%), Planctomycetes (3.5–9.6%), Euryarchaeota (0.2–10.9%), SAR406 (1.3–6.1%), and Cyanobacteria (0.8–6.2%) (Fig. 3b). Metagenomic sequences of Cyanobacteria (8.1%, 1.2%), α-Proteobacteria (28.8%, 9.3%), γ-Proteobacteria (47.9%, 40.3%), Euryarchaeota (0.7%, 1.5%), and Actinobacteria (0.9%, 2.5%) were also dominant at 30 m and 90 m in the blue hole, respectively (Fig. S2). In the blue hole, γ-Proteobacterial genus Alteromonas 16S rRNA sequences were abundant throughout the water column, especially at 30 m, 80 m, and 90 m (23.3–34.9%). Alteromonas species are widespread in shallow and deep waters of global oceans, including the ETNP OMZ55,56,57. Alteromonas species are particle-associated microaerophilic bacteria. In addition to relying on phytoplankton-derived organic matter for survival, Alteromonas species can also use NO3− as a nitrogen source58. SAR406 might participate in sulfur cycling via dissimilatory polysulfide reduction or sulfide oxidation59. The abundance of 16S rRNA sequences affiliated with SAR406 was 5.4–6.1% at 70–90 m in the blue hole, equivalent to SAR406 abundances at 150 m and 300 m at C3 and C4 (4.9–10.1%). SAR406 sequences were also highly abundant in the global OMZs46,59. In addition, 16S rRNA sequences affiliated with the methane-oxidizing archaean Marine Group II (phylum Euryarchaeota, class Thermoplasmata) were highly abundant in the blue hole at 70 m and 90 m (10.9% and 6.0%, respectively). These levels were comparable to Marine Group II abundance at 150–300 m at C3 (5.2–12.7%). The nitrite-oxidizing autotrophic Nitrospina (phylum Nitrospirae) was abundant between 50 m and 90 m in the blue hole (3.3–7.2%). The greatest Nitrospina abundance was at 90 m in the blue hole (7.2%), at 300 m at C3 (8.6%), and at 300 m at C4 (4.2%), suggesting that this genus occupied a wide range of niches.
Anoxic bottom layer
In the anoxic deeper waters of the blue hole (100–300 m), O2 was <1.0 µmol l−1, concentrations of H2S, NH4+, SiO32−, PO43−, and CH4 increased with depth, and only trace amounts of NO2− and NO3− were detected20. The microbial composition in this water layer was distinct, with the most abundant 16S rRNA amplicon sequences affiliated with the γ-Proteobacteria (11.9–42.2%), δ-Proteobacteria (12.0–18.7%), Candidatus OP3 (6.3–13.1%), Planctomycetes (2.0–9.3%), and Candidatus Parcubacteria (3.0–7.8%) (Fig. 3b). Also, metagenomic sequences of γ-Proteobacteria (67.4%) were dominant at the bottom waters of the blue hole (Fig. S2). The 16S rRNA amplicon sequences associated with the δ-Proteobacteria primarily included SO42− reducers, such as species from Desulfarculaceae, Desulfobulbaceae, and Desulphobacteraceae. We also identified 16S rRNA amplicon sequences affiliated with heterotrophic γ-Proteobacterial Pseudoalteromonas (29.6% at 200 m, 21.9% at 300 m) and Alteromonas (15.3% at 300 m, ~11.0% at 125–150 m), ε-Proteobacterial sulfur oxidizer Arcobacter (24.1% at 100 m), and phototrophic Prosthecochloris (Chlorobi, 7.2% at 100 m).
The O2-deficient environments often display ecologically specialized microbial populations, potentially mediating organic carbon turnover and syntrophic interactions. In the bottom layer waters of the blue hole, the clades of syntrophic taxa identified could potentially degrade lignocellulosic plant material or algal-derived complex organic polymers in order to produce hydrogen (H2), including Fibrobacter succinogenes (phylum Fibrobacteres)60, Latescibacteria61, and Firmicutes. The syntrophic bacteria also included taxa that convert small molecular compounds, such as glucose, pyruvic acid, short chain fatty acids, and glycerol to acetate and H2 for CH4 production—e.g., Thermotogae, Spirochaetae, Sebaldella termitidis (Fusobacteria), Elusimicrobium minutum (Elusimicrobia), Cloacimonetes, Atribacteria, Candidatus Acetothermus autotrophicum (Acetothermia), and Candidatus Hydrogenedentes62. Therefore, the blue hole represented a great amount phylogenetic and functional diversity of microbial communities that could drive matter and energy transformation throughout the water column.
Nitrogen-based metabolic potential
NH4+ production
NH4+ is a central component of the marine nitrogen cycle. Sources of marine NH4+ include the degradation of organic nitrogen compounds, ammonification, N2 fixation, hydrolysis of urea, and DNRA63. We identified genes encoding molybdenum-iron nitrogenase (MoFe, nifHDK) in the blue hole, affiliated with Cyanobacteria, Chlorobi, Bacteroidetes, Proteobacteria, Firmicutes, and Verrucomicrobia. The gene of nifH increased with depth, indicating that the microbial fixation of N2 was more common in deep waters of the blue hole (Fig. 4a). We also identified ureABC genes, which encode urease, associated with Thermoplasmata, Thaumarchaeota, Cyanobacteria, Actinobacteria, and Proteobacteria. High abundance of ureC gene at 10 m and 30 m (59.1% and 76.3%) was associated with the clades of Cyanobacteria and Alteromonas australica (γ-Proteobacteria) (Fig. 4b). The gene of nrfA, encoding dissimilatory ammonia-forming nitrite reductase, peaked at 100 m (4.5%), and was primarily detected in γ-Proteobacteria and δ-Proteobacteria (Fig. 4c).
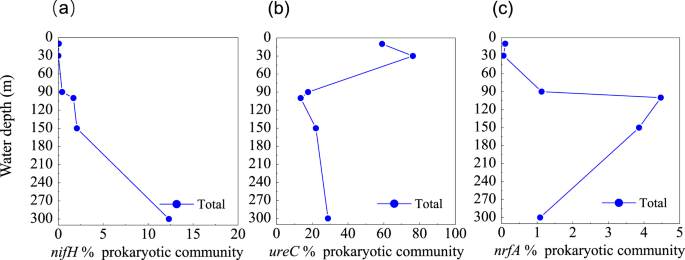
Profile of the abundances of (a) nifH, (b) ureC, and (c) nrfA genes for NH4+ production, from the Sansha Yongle Blue Hole. The abundance of functional genes was shown relative to the putative single copy per organism of RNA polymerase subunit B (rpoB). Abundances per gene are normalized to gene length.
Denitrification
The first step in denitrification—NO3− reduction to NO2−—can be catalyzed by nitrate reductases. The metagenomes in the blue hole were enriched in the narG gene, which encodes respiratory nitrate reductase, and the napA gene, which encodes periplasmic nitrate reductase at 90 m, accounting for 10.3% and 102.3% of prokaryotic community, respectively (Fig. 5a,b). This corresponded well with a reduction in NO3− concentration and the SNM at 90 m (Fig. 6d,g), implying NO3− reduction activity. More than 100% of the prokaryotic community contained the napA gene, implying multiple copies per genome in some members. At 90 m, the narG sequences primarily matched α-Proteobacteria, as well as γ-Proteobacteria (Enterobacteriaceae and a thioautotrophic gill symbiont of Bathymodiolus septemdierum). The proportion of narG gene was much higher at 150 m and 300 m than at 90 m, Alteromonadales and unclassified bacteria contributed to the high abundance, however, the capacity for these populations to perform NO3− reduction under trace NO3− and NO2− conditions is unknown. The napA gene sequences primarily matched γ-Proteobacteria (Aeromonas hydrophila, Thiolapillus, endosymbionts from an unidentified scaly snail isolate Monju, and Candidatus Thioglobus sp. EF1), ß-Proteobacteria (Sulfuricella denitrificans and Burkholderia xenovorans), and ε-Proteobacterial genus Arcobacter. In addition, napA gene from Alteromonas macleodii accounted for up to half of all napA gene sequences at 30 m, suggesting that these species might be responsible for the PMN formation (Fig. 5b).
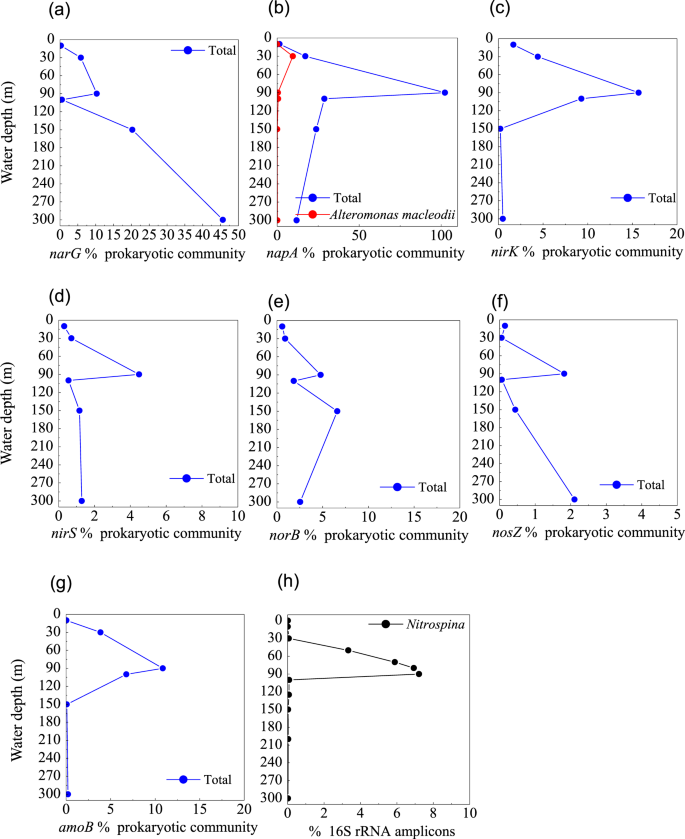
Profile of the abundances of (a) narG, (b) napA, and (c) nirK, (d) nirS, (e) norB, and (f) nosZ genes for denitrification, (g) amoB gene for nitrification, (h) the relative abundance of Alteromonas based on 16S rRNA amplicon sequences from the Sansha Yongle Blue Hole.
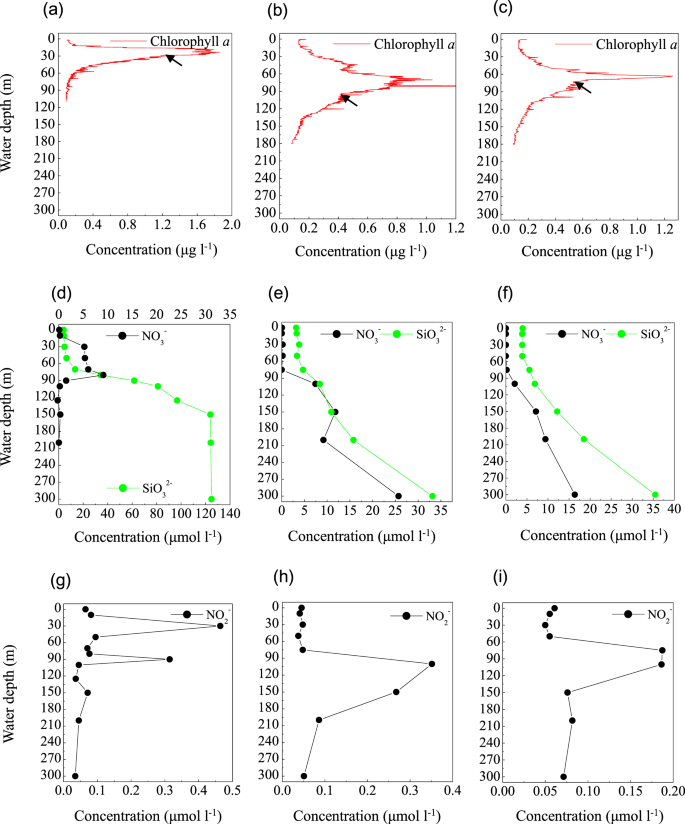
Chlorophyll a concentration, NO3− and SiO32− concentrations; NO2− concentration from the Sansha Yongle Blue Hole (a,d,g); C3 (b,e,h); C4 (c,f,i). The arrows indicate the onset of PNM. (d–i) were based on our parallel hydrochemical study20.
Genes that could accomplish other steps of denitrification were identified from a consortium of diverse members, indicating a high genomic potential for complete denitrification to N2 in the blue hole. These denitrification genes encoded copper-containing nitrite reductase (nirK), iron-containing nitrite reductase (nirS), nitric oxide reductase (norB), and nitrous oxide reductase (nosZ), but were not detected at high frequencies in comparison to narG and napA. NO2− reduction to NO is mediated by nirK/nirS, and the greatest number of nirK gene was detected at 90 m, where it was present in ∼16% of the prokaryotic community (Fig. 5c). Marine Group I Thaumarchaeota was the dominant nirK-containing population. The nirS gene was present in a lower percentage of the community than nirK, but were also most abundant at 90 m (4.5%) (Fig. 5d). NO reduction to N2O is mediated by norB, which was affiliated with γ- and ε-Proteobacteria in this study, achieving two maxima at 90 m and 150 m, respectively (Fig. 5e). N2 production from N2O is mediated by nosZ gene, which peaked at 90 m and 300 m in abundance (Fig. 5f). The Rhodospirillaceae-related nosZ was abundant at 90 m, corresponding well to the upper nosZ maximum, and Flavobacteriaceae-related nosZ was abundant at 300 m. Flavobacterial nosZ was also detected in marine O2-deficient zones in the ETNP64. N2O produced in shallow water is likely to be released into the atmosphere65. However, the N2O concentration from the surface layer of the blue hole was similar to open sea surface water20, suggesting that a new balance may have been established.
Nitrification
Ammonia monooxygenase catalyzes NO2− production via NH4+ oxidation. The amoB gene encoding ammonia monooxygenase was primarily associated with Nitrosopumilus (Thaumarchaeota), reaching a maximum of 10.9% of the prokaryotic community at 90 m in the blue hole (Fig. 5g). This maximum in amoB corresponded well with the SNM and the onset of the NH4+ increase, implying that NO2− accumulation occurs via NH4+ oxidation. The relative abundance of Nitrospina based on 16S rRNA amplicons in the blue hole, increased with depth (0–90 m) and in parallel with NO3− concentration, indicating that the NO2−-oxidizing chemoautotroph might produce the observed NO3− (Figs. 5h and 6d). Nitrospina was also the main driver of NO2− oxidation in the upwelling areas of the Eastern South Pacific, increasing in abundance with depth66,67. However, the nxr gene (encoding a nitrite-oxidizing enzyme, nitrite oxidoreductase) could not be detected in the metagenomes from the blue hole water column, suggesting a low abundance of Nitrospina. The relative abundance of Nitrospina might be overestimated based on measured 16S rRNA amplicons.
Anammox
To date, Scalindua is the only genus of anammox bacteria found in marine environments68. Low abundance of 16S rRNA amplicons matching Scalindua was present at the water depth between 80 m and 100 m (0.01–0.02%) in the blue hole, where NH4+ and NO2− overlapped at 80 m and 90 m, and NO2− began to disappear at 100 m. However, Scalindua-related sequences were not recovered in the metagenomes from the blue hole. This suggested that denitrification could be the dominant pathway of N2 formation in the blue hole, considerably outpacing anammox. The NO2− depletion in the bottom waters could limit the anammox pathways, although high NH4+ concentration was detected. Moreover, H2S could also inhibit the anammox activity69. This phenomenon was also detected in the OMZ off Peru in association with a giant H2S plume6. In contrast, abundant anammox activity was detected in the suboxic zone of the Black Sea where high levels of NO3− and NO2− were present69.
The primary NO2− maximum (PNM)
In the blue hole, based on the depth of the chlorophyll a peak base (~50 m) (Fig. 6a) and the onset of SiO32− accumulation (50 m) (Fig. 6d), we first hypothesized that the PNM would be located at ~50 m, consistent with the theoretical euphotic limit (51.2 m), and similar positions observed for the PNM in C3 (Fig. 6b,e,h) and C4 (Fig. 6c,f,i). Unexpectedly, the primary maxima of both NO2− and N2O were identified at 30 m, close to the depth of the chlorophyll a peak (Figs. 6g and 7e). At 30 m, we also identified peaks for a primary O2 minimum (~130 μmol l−1, Fig. 7a), a primary POC, and particulate nitrogen (PN) (Fig. 7d). However, the NO3− concentration peaked at the bottom of the PNM (80 m, Fig. 6d). These are all classic signals for denitrification. Indeed, Alteromonas species were maximally abundant (34.9%) at 30 m in the blue hole (Fig. 7b). Of these species, one particularly abundant species (32.1%) with a 99% identity to Alteromonas macleodii was identified (Fig. S3a). Moreover, the NapA gene from Alteromonas macleodii accounted for up to half of all napA gene sequences at 30 m (Fig. 5b). Therefore, we speculated that, at 30 m in the blue hole, photoautotrophs formed large amounts of POC, which fueled microbial growth and aerobic respiration, leading to O2 deficency. In addition, phytoplankton particles generate microscale oxyclines for suboxic or anoxic respiration in oxygenated waters57. Based on the formula of Stief et al.70, given an ambient O2 of 130 µmol l−1 at 14 °C, O2 concentration at the center of the diatom aggregate was ~40 µmol l−1, comparable to the value of 39 µmol l−1 that inhibits NO3− reduction71. Reasoning that the O2 solubility decreases with increasing temperature, at higher temperature of 25.6 °C at 30 m20, O2 concentration would be even lower within the organic aggregates. At such low O2 concentration, Alteromonas species might reduce NO3−, leading to the accumulation of NO2− in oxygenated waters. Experimental conditions have measured NO3− reduction at low O2 concentrations, which presumably matches to anoxic micro-environments71,72. Isolating and culturing an Alteromonas macleodii strain from 30 m in the blue hole revealed that this strain grew statically in diluted liquid 2216E marine medium (0.5 g yeast, 2.5 g tryptone, 1-L sea water) supplemented with 300 µmol l−1 NaNO3 for 3 d, and NO2− accumulation was evident (5.6 µmol l−1, unpublished data). This suggested that Alteromonas macleodii could perform NO3− reduction in the stagnant water. Altogether, in the O2-limited blue hole, a PNM at shallow water depth was identified and the denitrification activity of Alteromonas species might play important role in generating the PNM. Additionally, denitrification by aggregate-associated bacteria may shift the PNM towards the chlorophyll a peak in an O2-deficient marine system, which may previously have been overlooked. In addition, a primary NH4+ maximum was detected between 20 m and 80 m in the blue hole20. Low abundance of amoB gene sequences coupled with NH4+ substrate at 30 m could also partly contribute to NO2− accumulation (Fig. 5g).
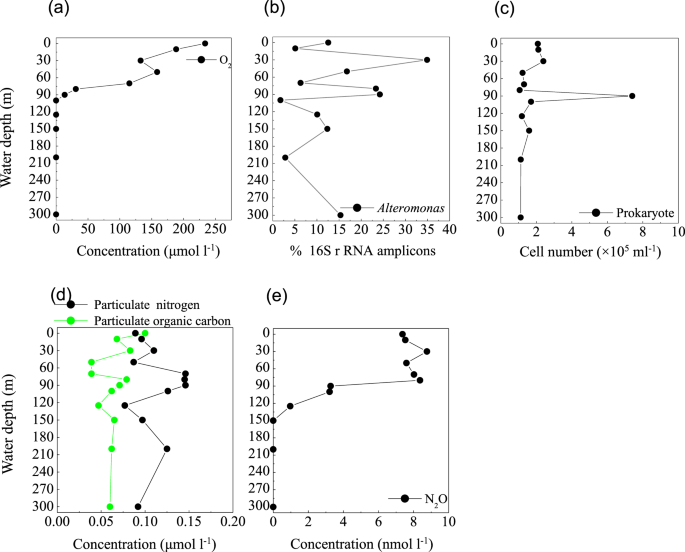
Profile of (a) O2 concentration, (b) the relative abundance of Alteromonas based on 16S rRNA amplicon sequences, (c) prokaryotic cell number, (d) Particulate nitrogen and particulate organic carbon concentrations, and (e) N2O concentration from the Sansha Yongle Blue Hole. (a,d,e) were based on our parallel hydrochemical study20.
The secondary NO2− maximum (SNM)
In low O2 environments, a SNM is often detected below the PNM73. NO2− in the SNM is mainly produced by dissimilatory NO3− reduction, an alternative respiratory mechanism that becomes favorable when O2 is limited. We observed a SNM in the blue hole at 90 m in close proximity to the base of the NO3− maximum (where O2 concentration had decreased to 13.4 µmol l−1) (Figs. 6g and 7a). The maximal POC and PN concentrations occurred in this layer, as well as highest abundance of prokaryotes (Fig. 7d,c). Both narG and napA genes present in heterotrophic Proteobacteria were also enriched at 90 m in the blue hole (Fig. 5a,b). Thus, at 90 m in the blue hole, O2-deficient condition and a high particle load might lead to an alternative respiration prevalent, with NO3− as an electron acceptor. In addition, at 90 m, populations containing NO3− reducing genes also harbored sulfur-oxidizing genes, including γ- Proteobacteria (thioautotrophic gill symbiont of Bathymodiolus septemdierum, and Candidatus Thioglobus), ε-Proteobacteria (Arcobacter and Sulfurimonas), and Chlorobiaceae. Therefore, the sulfur-driven chemolithotrophic denitrification could also be a crucial method for SNM formation. In addition, amoB gene reached a maximum of 10.9% of the community at 90 m, which might also be partly responsible for the NO2− accumulation (Fig. 5g).
Sulfur-based metabolic potential
Sulfate reduction
Under O2 depletion, both episodic plumes of H2S in continental shelf regions and permanent H2S under sulfidic conditions are produced by SO42–-reducing bacteria from SO42– 4,6,7. Based on 16S rRNA amplicons, diverse SO42−-reducing populations were detected at 90 m, accounting for 0.4% of total prokaryotes in the blue hole, which increased rapidly between 100 m and 300 m (10.6–16.7%). These SO42−reducers included Desulfatiglans (family Desulfarculaceae, 3.0–10.2%) and Desulfurivibrio (family Desulfobulbaceae, 0.1–2.6%). In addition, an unclassified genus in the Desulfobacteraceae (2.4–3.9%) and an unclassified genus in the Desulfobulbaceae (0.4–5.8%) were also identified. Among these taxa, Desulfococcus (0.04–0.21%) and Desulfovibrio (0.05–0.21%) were also detected in OMZ waters off the Chilean Coast4. The relative abundances of sequences associated with the Desulfovibrionaceae, Desulfarculaceae, Desulfobulbaceae, and Desulphobacteraceae were represented in Fig. 8a. In good agreement with this data, metagenomic results suggested that gene sequences encoding dissimilatory sulfite reductase (dsrA) were present in high proportions between 90 m and 300 m (1.0–9.3% of the community) (Fig. 8b). In contrast, SO42−-reducing population represented only ~0.04% between 0 m and 80 m in the blue hole, and 0.1–0.2% in the surrounding regions. The dsrA distribution was paralleled by SO42−-reducing populations and the H2S concentration (Fig. 7c) in the blue hole. Therefore, SO42− reduction in the water column is an important pathway, and might contribute to large volumes of H2S, creating a sulfidic zone as thick as ~200 m.
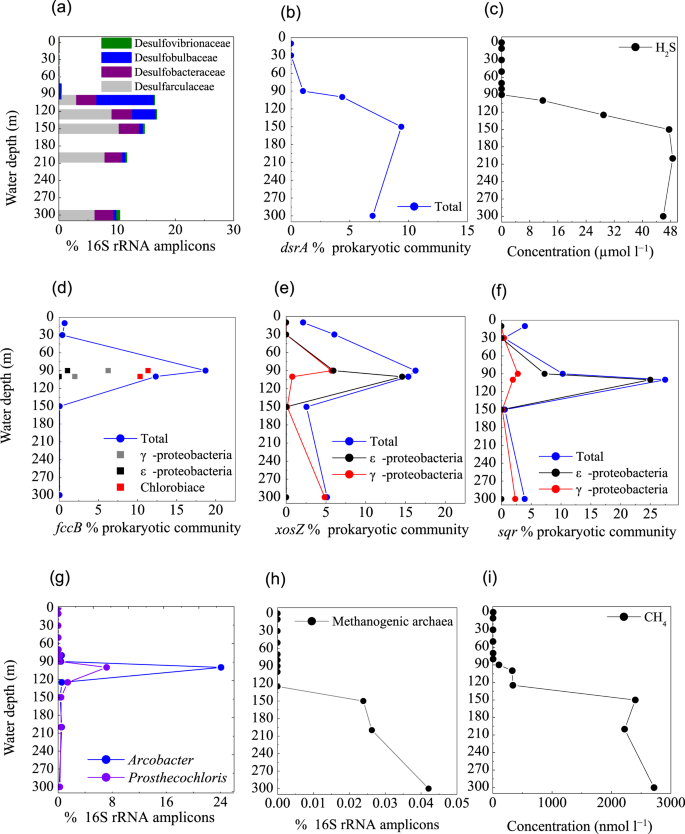
Profile of the abundances of (a) representative SO42−-reducing populations, (b) dsrA gene for sulfate reduction, (c) H2S concentration, (d) fccB, (e) soxZ, and (f) sqr genes for sulfur oxidization, (g) representative sulfur-oxidizing denitrifiers, (h) Methanogenic archaea, (i) CH4 concentration, from the Sansha Yongle Blue Hole. (c,i) were based on our parallel hydrochemical study20.
Sulfur oxidization
Clades of sulfur-oxidizing bacteria are particularly enriched at the oxic–anoxic interfaces, where O2, NO3−, and metal oxides are available as electron acceptors6,7,19,74,75,76. At these interfaces, H2S can be oxidized using the sulfide: quinone oxidoreductase enzyme (Sqr) and flavocytochrome c/sulfide dehydrogenases (Fcc), forming SO3−. The SO3− can be further oxidized to SO42− by the adenylylsulfate reductase (Apr) and sulfate adenylyltransferase (Sat). Elemental sulfur and S2O32− are presumably oxidized to SO42− via the sulfur-oxidizing multienzyme complexes (Sox)77.
Mining the metagenomic data, we identified genes that could allow for dissimilatory sulfur oxidation, including genes that encode sulfide: quinone-oxidoreductase (sqr), flavocytochrome c/sulfide dehydrogenase (fccABC), sulfate adenylyltransferase (sat), adenylylsulfate reductase (apr) and sulfur-oxidizing multienzyme complexes (soxABCD and soxYZ) in the blue hole. These genes were detected in various combinations across diverse sulfur-utilizing taxa, primarily affiliated with γ-, ε-Proteobacteria, and Chlorobi. The greatest abundances of fccB and soxZ genes were present at the suboxic layer (90 m) (Fig. 8d,e), while sqr gene was present at the upper anoxic layer (100 m) (Fig. 8f), coinciding with a steep decline in H2S concentration20. This suggested that H2S oxidation may occur at the NO3−/NO2−-H2S transition in the blue hole. Further, fccB gene affiliated with Chlorobiaceae was comparable at 90 m and 100 m (~10% of community) (Fig. 8d). Genes encoding fccB, sqr, and soxZ affiliated with Candidatus Thioglobus (γ-Proteobacteria SUP05) were dominant at 90 m, while genes of sqr and soxZ affiliated with ε-Proteobacterial genera Sulfurimonas and Arcobacter were enriched at 100 m (Fig. 8d–f). Depth-specific patterns among different sulfur-metabolizing taxa might reflect differences in O2 sensitivity, as well as adaptations to varying energy substrates.
Chlorobiaceae species are phototrophic bacteria78. The high abundance of fccB gene(11.4% at 90 m and 10.3% at 100 m) and 16S rRNA amplicons (7.2% at 100 m), within the narrow layer might indicate that Chlorobiaceae members could potentially couple H2S oxidation to phototrophy, even at extremely low-light intensities (Fig. 8d,g). The Chlorobiaceae taxa contained sequences encoding nitrate reductase and nitrous reductase, as well as a RuBisCO-like protein for CO2 fixation. This suggested Chlorobiaceae could use NO3− as a potential terminal electron acceptor for H2S oxidation, linked to CO2 assimilation via Calvin cycle for dark primary production.
The metagenomic data suggested that sulfur oxidizing genes found in Candidatus Thioglobus (SUP05) were enriched in the suboxic and anoxic zones of the blue hole. Genes of sqr (0.7% at 90 m, 0.2% at 100 m), fccB (6% at 90 m, 1.4% at 100 m), soxZ (5.0% at 90 m) and napA (0.03% at 90 m, 0.01% at 100 m) were recovered, implying that Candidatus Thioglobus may prefer to reside within suboxic zone. These results support finding from recent surveys indicate that γ-Proteobacterial SUP05 can oxidize sulfur by denitrification, and is most abundant in slight to moderate redoxclines, thereby linking sulfur cycling to N-loss pathways4,12,37,38.
In contrast to SUP05-related sequences, ε-Proteobacteria preferentially colonized anoxic and highly sulfidic environments in the blue hole. The soxZ gene related to Sulfurimonas occupied 13.8% of the community at 100 m, although a minor component of the 16S rRNA amplicons (0.4%) affiliated with Sulfurimonas was detected. In addition, up to half of the norB sequences were related to Sulfurimonas species, further suggesting that the sulfur-oxidizing genus Sulfurimonas also supported reductive nitrogen metabolism. Sulfurimonas species are also widespread in the sulfidic anoxic waters of the Benguela system off Namibia7, as well as in the anoxic waters of the Baltic Sea, the Black Sea41, and the Cariaco Basin42. The sqr sequences were present in high abundance in Arcobacter (7.2% at 90 m and 25.0% at 100 m). Correspondingly, 16S rRNA amplicons affiliated with Arcobacter were also most abundant at 100 m (24.1%) (Fig. 8g). Based on the alignment of these 16S rRNA sequences with previously published sequences in GenBank, one Arcobacter-affiliated sequence from the blue hole (23.9%) were 99% identical to gill epibionts of hydrothermal vent gastropods, and Arcobacter clones from the Saanich Inlet, from the near-shore anoxic basin, and from the costal oxycline, respectively. Another Arcobacter-affiliated sequence also had 96% identity to Arcobacter nitrofigilis and 95% identity to Arcobacter sulfidicus (Fig. S3b). Arcobacter-associated sequences were also found in the OMZs off Peru6 and the sulfidic Benguela system off Namibia7, accounting for ~2–10% in abundance. These species were identified as key organisms in the chemolithotrophic oxidation of H2S with NO3−7. The metagenomic data from our study indicated that Arcobacter species might perform denitrification (napA, nir, nor), as well as oxidizing HS–/S2– to S0 (sqr, fccB) and SO32− to SO42− (soxACD and soxYZ) for energy generation. Additionally, Arcobacter-affiliated sequences contained genes encoding clades of proteases, peptidases, and oligopeptidases, as well as enzymes critical for the oxidative tricarboxylic acid (TCA) cycle (citrate synthase). However, no glycerases were identified. This indicated that Arcobacter species used proteins, amino acids, propionates, and TCA cycle intermediates, but not carbohydrates79. We also identified gene sequences encoding key enzymes of the rTCA cycle for chemoautotrophic CO2 fixation, including citrate lyase (aclB), pyruvate flavodoxin oxidoreductase (porA), and 2-oxoglutarate-acceptor oxidoreductase (oorA). Therefore, Arcobacter species had the genomic capacity to grow chemolithoautotrophically via H2S or S2O32− oxidation that is linked to diverse steps of denitrification, as well as heterotrophically on various organic compounds. The metabolic versatility of Arcobacter might provide a competitive advantage in the energy-limited blue hole.
The microbial reduction of NOx coupling to sulfur oxidation pathways has been documented in diverse taxa from the H2S/NO3− transition zones in OMZs4,6,35,36,37,38,39,40,41. In the blue hole, sulfur-oxidizing denitrifiers—such as γ-, ε- Proteobacteria, and Chlorobiaceae—were enriched at 90 m and 100 m, supporting sulfur oxidation that is coupled to reductive nitrogen metabolism. It is obvious that sulfur-based denitrification occurs in this zone (90 m), where NO3−/NO2− and H2S overlapped. Meanwhile, amoB gene from Nitrosopumilus (Thaumarchaeota) was recovered at 100 m, indicating that NH4+ oxidation could provide the NO2− substrate necessary for denitrification, although this process is transient and cryptic, as trace NO3−/NO2− was detected at 100 m. This is in good agreement with a previous report on the anoxic water at Landsort Deep of the Baltic Sea47. We speculate that H2S produced by heterotrophic sulfur reducers could support sulfur-driven chemolithotrophic denitrification, which mediates both nitrogen loss and H2S removal from the blue hole.
CH4 cycle
In the blue hole, sequences associated with methanogens (phylum Euryarchaeota, order Methanomicrobiales and Methanosarcinales) were identified at 150–300 m, with a total abundance of 0.02–0.04% of total 16S rRNA amplicons (Fig. 8h). The total abundance of these taxa at 150–300 m was linear positively correlated with the concentration of CH4 (~2.4–2.7 µmol l−1, Fig. 8i, r = 0.838). Gene encoding methyl-coenzyme M reductase (mcrA), the best diagnostic enzyme for anaerobic methanogenesis, was not found in the metagenomic data. This could be explained by low levels of archaeal 16S rRNA amplicons. However, metagenomic and metatranscriptomic data in the 300 m surface sediment revealed a mcrA gene belonging to Methanosarcinales, suggesting active methanogenesis (unpublished data). Based on this study’s 16S rRNA amplicons and metagenomic sequences, coupled with recent published literatures, we propose three methanogenic pathways in the bottom waters of the blue hole. (1) Methanococcoides and Methermicoccus adopt methylotrophic pathways, including one-carbon compound pathways such as methanol conversion to CH480. Consistently, gene sequences for key enzymes were found, such as trimethylamine-corrinoid protein Co-methyltransferase and Methylated-thiol-coenzyme M methyltransferase. (2) Methanosaeta and Methanosarcina catalyze the acetoclastic pathway (acetate conversion to CH4)81,82. (3) The family Methanomicrobiaceae catalyzes the hydrogenotrophic pathway (H2 + CO2 → CH4)83. In terms of abundance, methylotrophic methanogenesis was the major pathway in the blue hole, in agreement with previous reports that some methanogens can survive in the presence of SO42- reducers by consuming noncompetitive methylated substrates84. In contrast, SO42− reduction processes could compete for these substrates, (e.g., H2 and acetate), potentially leading to a low proportion of sequences related to hydrogenotrophic and acetoclastic methanogenesis. Sequences affiliated with CH4-oxidizing archaeal Thermoplasmata displayed comparable abundance among the blue hole and the open sea waters, potentially explaining the low concentration of CH4 (<9 nmol l−1) in the oxic layers.
Source: Ecology - nature.com

