Simulation overview
All simulations take place in a one-dimensional circular array, occupied by a specified number of trees (n), each with a unique community of root-associated microbial pathogens and mutualists. We chose to conduct the simulations in one dimension to simplify interpretation, but we recognize that simulation dynamics likely progress more slowly than they would in two dimensions50. At each time step, the simulation randomly selects a subset of the tree population for replacement. All trees produce a specified number of seeds at each time step that disperse according to a power-law dispersal kernel. The probability that an individual of a given tree species recruits to the adult population at a site is a function of the community of microbial propagules (pathogens and mutualists), and the relative abundance of the tree species’ seeds, occurring at the site. A microbial community is then calculated for the recruit based on the abundances of the microbes at its location, the compatibility between the microbes and the species of recruit, and the competitive ability of the microbes. At each time step, the populations of microbes change on each adult according to competition equations defined below. The simulation continues for an arbitrary number of time steps.
Assumptions
- 1.
All plant species are equivalent, except in the identity of their microbial pathogens and mutualists, and the absolute survival probability of their seedlings (a representation of fitness differences that is unrelated to interactions with microbes). In the absence of plant-microbial interactions, plant communities develop according to neutral expectations.
- 2.
Plants, mutualists, and pathogens are all dispersal limited, and follow a power-law dispersal function. We allow the scale parameter (indicating dispersal distance) of the dispersal kernel to vary between plants, pathogens, and mutualists, respectively.
- 3.
Microbial taxa (both pathogens and mutualists) have two host compatibility levels: one value for their primary host, and another value—less than or equal to the value for the primary host—for all other host species. This means that all non-target hosts are equivalent for each microbial taxon. Thus, in the simulation, pathogens and mutualists can be complete host specialists, complete generalists, or capable of associating with all hosts to varying degrees. Empirical results suggest that many pathogens and mutualists may associate with a broad range of hosts, but that they have host-specific effects32.
- 4.
Mutualists are beneficial, and pathogens are detrimental to host survival.
- 5.
Each microbial taxon has an identical carrying capacity on each host.
- 6.
The effect of a microbial taxon on its host is correlated with the competitive ability of the microbial taxon on that host (i.e. more beneficial mutualists outcompete less beneficial mutualists, and more detrimental pathogens outcompete less detrimental pathogens)51,52,53.
- 7.
Once a tree becomes an adult, its microbial community is subject to change according to internal dynamics, and immigration of microbial propagules from nearby trees. We choose to model these changes according to competition equations with logistic growth.
Simulation description
Each simulation starts as a circular array populated with n trees, of k species, evenly positioned through space. Each tree species is the preferred host for one mutualist species and one pathogen species, but concomitantly has the potential to associate with all pathogens and mutualists to varying degrees defined by the simulation. Thus, the mutualist and pathogen community associating with each tree is described by a vector of abundances.
For each microbial taxon, the degree of affinity, s, ranges from zero (complete specialist that only generates an effect on preferred hosts) to one (complete generalist that generates an equal effect on all hosts). All microbial species in a given guild, f (mutualists, m, and pathogens, p), have a guild-specific specificity value, sf, such that the compatibility of a microbe i with host species j is:
$$e_{fij} = left{ {begin{array}{*{20}{c}} {1,, j = {mathrm{preferred}},{mathrm{host}}} {s_f,,j , ne , {mathrm{preferred}},{mathrm{host}}} end{array}} right.$$
(1)
The probability that the seedling survives to recruit successfully to the adult population is determined by the microbial community at the location to which it disperses, and the compatibility between the microbial taxon and the candidate seedling. The microbial community at any given location, Fx (mutualists, Mx, or pathogens, Px) is the sum of the microbial abundances occupying the adult in the cell at time t, and the microbes arriving as propagules. The density of pathogen and mutualist propagules at a given location are determined by taking the sum of the microbial communities of all adults, scaled by their distance from the location according to a truncated discrete power-law distribution:
$${boldsymbol{F}}_{boldsymbol{x}}(t + 1) = {boldsymbol{F}}_{boldsymbol{x}}(t) + gamma _fmathop {sum }limits_{j = 1:n} {boldsymbol{F}}_{h_j}(t) cdot {mathrm{powerlaw}}left( {left| {x – {mathrm{pos}}left( {h_j} right)} right|} right)$$
(2a)
$${mathrm{powerlaw}}left( x right) = frac{{(1 + x)^{ – b_f}}}{{mathop {sum }nolimits_{n = 0}^infty (1 + n)^{ – b_f}}}$$
(2b)
where pos(hj) corresponds to the position of host j, and γf represents the fecundity of the mutualists or pathogens. The mutualist community, Mx, and the pathogen community, Px, are used to calculate the recruitment probability of the seedling. At each recruitment trial, this compatibility score is used to calculate the impact of a given microbial taxon on the probability of recruitment of its host. For mutualists and pathogens, the impact is equal to the sum of the abundance of each microbial taxon multiplied by its compatibility with the focal host. So, for a given seedling of species j, at location x, the effect of the microbial community on the seedling is:
$$theta _fleft( {x,j} right) = {boldsymbol{F}}_x cdot {mathbf{e}}_{fj}$$
(3)
where Fx represents the vector of microbial abundances at location x, and ({mathbf{e}}_{fj}) represents the vector of effect values of microbes on species j. The total effects of pathogens and mutualists determine the recruitment probability, Π, of a seedling via the following function:
$$begin{array}{l}{Pi}left( {theta _{mathrm{m}},theta _{mathrm{p}},j} right) = frac{{{zeta}_j{mathrm{logistic}}left( {gleft( {htheta _{mathrm{m}} – theta _p} right)} right)}}{{mathop {sum}nolimits_{n = 1:k} {{Pi}left( {theta _{mathrm{m}},theta _{mathrm{p}},n} right)} }} quad quad quad quad quad {mathrm{logistic}}(x) = frac{{1}}{{1 – {mathrm{e}}^{ – x}}}end{array}$$
(4)
where ζj represents the relative fitness of plant species j. The coefficient derived from Eq. (4) is calculated for each plant species arriving at an empty cell, and standardized by the number of seeds of each species landing in the cell. The successful recruit is then drawn from the resulting multinomial distribution.
The recruit becomes a reproductive adult in the next time step, and begins modifying the abundance of each microbial taxon through time according to the following differential equation:
$$begin{array}{*{20}{c}} {frac{{dF_{ij}}}{{dt}} = r_fe_{fj}^{q_f}F_{ij}left( {1 – left( {F_{ij} + mathop {sum}limits_{n ne j} {alpha _fe_{fn}^{c_f}F_{in}} } right)} right)} end{array}$$
(5)
For each microbial guild, rf represents the intrinsic growth rate when associating with its preferred host. When associating with a non-preferred host, we multiply by the effect of the microbe on non-target hosts, sf, raised to the exponent qf. The parameter αf represents the competitive ability of a microbial taxon when associating with its preferred host. The competition coefficient is multiplied by (s_f^{c_f}) when the microbe is associated with a non-preferred host. The microbial community changes on each tree in the manner described by Eq. (5) at each time step. This cycle of mortality and replacement continues for a predetermined number of time steps.
Measuring plant-soil feedback
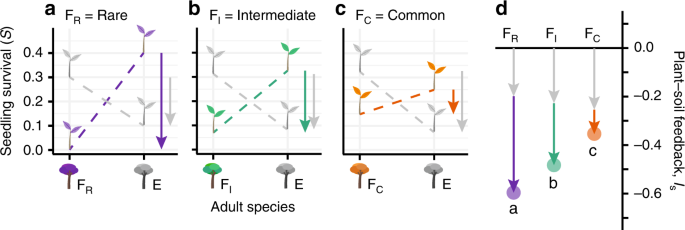
To offer results that are directly comparable to empirical studies, we tracked plant-soil feedback, the process that drives negative density dependence. We measured plant-soil feedback strength using Bever’s interaction coefficient, Is (See Supplementary Fig. 1)19,47. The metric can be calculated for any two plant species, A and B, as follows:
$$begin{array}{*{20}{c}} {I_{mathrm{s}} = Sleft( {a_{mathrm{A}}} right) + Sleft( {b_{mathrm{B}}} right) – left( {Sleft( {a_{mathrm{B}}} right) + Sleft( {b_{mathrm{A}}} right)} right)} end{array}$$
(6)
where (Sleft( {a_{mathrm{A}}} right)) and (Sleft( {b_{mathrm{B}}} right)) are the respective mean survival probabilities of seedlings of species ({mathrm{A}}) and ({mathrm{B}}) grown beneath conspecific adults (home performance), and (Sleft( {a_{mathrm{B}}} right)) and (Sleft( {b_{mathrm{A}}} right)) are the mean survival probabilities of seedlings of species ({mathrm{A}}) and ({mathrm{B}}) grown beneath adults of the other species (away performance). A given plant species’ value for Is is the mean of all pairwise feedback values that include the focal species. When using Is as a predictor of model outcomes (e.g. for random forest models), we measured Is at the beginning of the simulation. Specifically, we measured Is after allowing microbial communities to change on their hosts following Eq. (5) for 50 time steps without host mortality. To quantify the relationship between host abundance and PSF (at the end of each simulation run), we calculated the Spearman correlation coefficient between PSF and host abundance.
Determining whether simulations reached equilibrium
To establish whether plant abundances reached a stable equilibrium at the end of the simulation, we employed an equilibrium metric, Pe. To calculate Pe, we measured the change in abundance of the most common plant species across all windows of 100 time steps in the 600 time steps leading up to the end of the simulation (subsampled to include every 10th time step in order to improve computational speed). We then used a t-test to determine whether the average rate of change across 100 time steps deviated from zero. The equilibrium metric, Pe, is the P-value of this two-sided t-test. Higher values of Pe indicate that the simulation is more likely to have been at a stable equilibrium over the final 600 time steps.
Characterizing simulation behaviour
Given the large number of possible parameter value combinations, we used a random search to explore parameter space, then characterized model behaviour using a random forest classification approach30. For each run, parameter values were drawn from independent random uniform distributions defined in Table 1.
Random forest classification was used to construct models predicting whether (1) all plant species coexisted throughout the simulation, (2) whether strong negative feedback developed (i.e. the maximum strength feedback was less than −1.5), and (3) whether a strong positive correlation between host abundance and plant-soil feedback developed (ρ(Is) > 0.8, σ(Is) > 0.02, Imax < 0, and richness of plants, mutualists, and pathogens = 5). For each model, all parameter values were included as predictors. For model (1), we also included maximum PSF strength, Imax, as a predictor. Sample sizes for random forest fitting were balanced, such that both classes were equally represented. Hyperparameters for each random forest fit were otherwise set to the default settings for classification using the R package randomForest54.
For each random forest model, we estimated the importance of each parameter as a predictor of model output. We quantified variable importance as the decrease in prediction accuracy resulting from sample-wise permutation of the values among out-of-bag samples. I.e. if removal of a parameter from the classification model decreased the model’s prediction accuracy, the parameter was deemed important. Feature contributions of each model parameter were visualized as the out-of-bag cross-validated conditional contributions of each parameter value to the predicted model outcome55. In feature contribution plots, each point represents the average (among decision trees) contribution (y) of a parameter value (x) to the predicted outcome. Random forest analyses were conducted using the R package randomForest54, and the variable importance and contributions were estimated using the R package forestFloor55.
Simulation runs
We ran 16,000 simulations, each with five species of plants comprising 499 individuals. Parameter values were drawn randomly from the ranges listed in Table 1. For each simulation run, the starting location of each individual tree was randomized, and each tree and microbial species started at equal total abundances. At each time step, ten percent of the adult trees were replaced (equivalent to a 5-year time step, assuming two percent mortality yr−1). Simulations were run for 3 K total steps (equivalent to 15 K years). See Figs. 2 and 3 for an overview of simulation output.
Optimizing for correlation between abundance and feedback
The probability of randomly achieving a simulation parameterization that matched any set of PSF patterns was low, because we explored a very broad range of parameter values. This broad search allowed us to clearly understand the independent influence of each parameter on simulation results using random forest models, but also meant that any specific simulation result was poorly replicated in our random search. We therefore used particle swarm optimization (PSO) to identify combinations of parameter values most likely to simultaneously promote species coexistence under negative PSF, and generate a positive correlation between PSF and plant abundance (i.e. ρ(Is) > 0.8, σ(Is) > 0.02, Imax < 0, and richness of plants, mutualists, and pathogens = 5).
In PSO, combinations of parameter values are described as particles, and the population of particles is called a swarm. To update particle positions at each iteration, t, we first employed the following equation to determine particle velocity, v, of each particle (x_i) in each dimension d:
$$begin{array}{*{20}{c}} {v_{id}left( {t + 1} right) = w cdot v_{i,d}left( t right) + c cdot {mathrm{rand}} cdot left( {p_{id} – x_{id}} right)} end{array}$$
(7)
where w and c are constants (we selected 0.5, and 0.8, respectively), rand represents a random number drawn from a uniform distribution bounded by 0 and 1, and p represents the position of the best solution. At each step, we trained a new random forest classification model, then took the 10 solutions deemed most likely to produce negative feedback and the feedback-abundance correlation. For each particle in the swarm, we randomly selected one of the 10 best solutions to determine (p_{id}), then updated the position of each particle was determined with the following equation:
$$x_{id}left( {t + 1} right) = x_{id}left( t right) + v_{id}left( {t + 1} right)$$
(8)
At each time step, we randomly selected four particles to replace with random parameterizations to limit premature convergence. To ensure that we were not identifying a local optimum, we iteratively conducted PSF, starting with 16 non-overlapping optimizations. For each optimization, we allowed a swarm of 50 particles to iteratively explore the parameter space for 20 PSO steps, starting from random positions. We then combined all results from two optimization runs (i.e. eight combinations of 1000 total runs), and conducted eight PSO optimizations for another 20 time-steps. We continued this process until the final optimization included all simulation runs. This optimization process allowed us to confirm that independent optimizations identified similar solutions, rather than different local optima. We then used a final random forest classification model to identify the parameterization most likely to produce negative feedback and the feedback-abundance correlation among all runs conducted throughout the PSO optimizations (Figs. 2–4).
Isolating effects of model components on population dynamics
To characterize the importance of mutualists and pathogens in determining plant community dynamics, we conducted additional runs in which we removed the effect of (1) all microbes, (2) only pathogens, and (3) only mutualists, while tracking plant abundances through time. Specifically, after letting the simulation run for an equivalent of 4 K years, we removed each of the following model components from independent batches of 16 runs: (1) the effect of microbes on seedling survival (i.e. to simulate neutral dynamics); (2) only the effect of pathogens; (3) only the effect of mutualists.
For an additional batch of 16 runs, we fixed the microbial community composition on each plant species. At the simulation time step equivalent to 4 K years, we calculated the mean abundances of each microbial taxon on individuals of each host plant species. These species-specific average microbial communities were subsequently assigned to all new recruits for the rest of the simulation. This latter approach is analogous to the approach used in simulations that assume trees have a fixed effect on the survival of conspecific vs. heterospecific seedlings5,8,19.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Source: Ecology - nature.com
