Site description and soil property measurements
The soil warming experiment was set up at the Kessler Atmospheric and Ecological Field Station in McClain County, OK (34° 59′ N, 96° 31′ W). The mean annual temperature is 16.3 °C and mean annual precipitation is 914 mm [20]. The experiment was a blocked split-plot design and initiated in July, 2009. For each selected block, soil was sampled yearly through 2016 from three randomly selected warming replicates heated by above ground infrared radiators and from three paired ambient control plots located 5 m away. The sampling scheme, soil properties measurements, and DNA extraction from 48 soil samples used (2 treatments, 3 replicates over 8 years) were reported previously [15, 21].
Constructing Cas1 HMMs and Xander packages
The three files for automatically generating a Xander package include (1) a fasta file containing seed sequences; (2) an HMM, and (3) a fasta file containing a more diverse collection of the targeted protein sequences (framebot.fa). Near full length of cas1 protein sequences were retrieved from GenBank [22] for the well-curated protein family seed sequences in Pfam [23] (Pfam 01867), TIGERFAM [24] (TIGRFAM00287, TIGRFAM03637, TIGRFAM03638, TIGRFAM03639, TIGRFAM03640, TIGRFAM03641, TIGRFAM03983, TIGRFAM04093, and TIGRFAM04329) and the recent literature [2, 25,26,27,28,29]. The compiled cas1 protein sequences were aligned by MAFFT [30] in Jalview (jalview.org, v. 2.10.2b2). The aligned cas1 protein sequences were dereplicated to remove the identical or substring of sequences and clustered by sequence similarity using RDPTools (ReadSeq.jar and Clustering.jar; https://github.com/rdpstaff/RDPTools). The dereplicated cas1 protein sequences grouped into seven complete-linkage clusters at 50% identity cutoff. The sequences in each cluster were used as the seed sequences to build HMMs using modified HMMER 3.0 [31] with a patch file [10]. An additional HMM was added specifically for archaeal Type II cas1 considering its novelty [2]. To prepare the third file required for a Xander package (framebot.fa), we used the respective HMM to search against the nonredundant protein sequence database (nr, NCBI) via hmmsearch [31] and collected sequences annotated as ‘Cas1’. Eight cas1 Xander packages named as M1–M8 were automatically prepared using a shell script (https://github.com/rdpstaff/Xander_assembler/blob/master/bin/).
Evaluating and improving HMM performance
We created a reference genome set including 91 bacterial and archaeal genomes across 11 phyla carrying 93 cas1 sequences of 17 subtypes from NCBI Genome database (https://www.ncbi.nlm.nih.gov/genome) (Supplementary T1) to evaluate and train the newly constructed HMMs (Fig. 1b). The subtypes included Archaeal Type II, CasX, CasY, IA-IF, IU, IIA-IIC, and IIIA-IIID, which are classified followed the naming standard that has been specified by Makarova KS and Koonin EV [5]. Pairwise distances among the 93 cas1 protein sequences were calculated (https://github.com/rdpstaff/Clustering), and a cas1 protein tree was built by FastTree [32]. Together with the reference genome set, a genome of Streptomyces coelicolor (NC_003888.3) with a gene encoding transposase, a homolog of cas1 protein [33], was included as the outgroup to root the cas1 protein tree and also used to detect potential false positives in the following evaluation step.
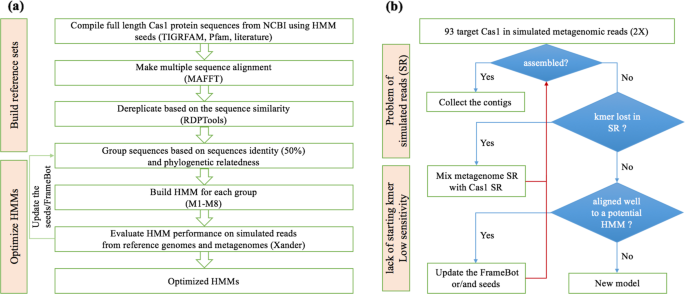
HMM construction (a): near full length of cas1 protein sequences previously used in Cas1 TIGRFAM, Pfam and mentioned in the literature were retrieved from NCBI and were cluster at 50% sequence identity after aligned by MAFFT and dereplicated by RDPTools. Eight HMMs were constructed based on the seed sequences in each cluster. Model optimization (b): the HMM performance was evaluated by the simulated reads generated from a set of reference genomes carrying 17 subtypes of CRISPR-Cas systems. We optimized the HMMs by updating the coverage of the corresponding seed sequences and Framebot files.
First, we used the reference genome set to evaluate the sensitivity and specificity of the eight HMMs. Simulated metagenomic reads from the 91 genomes were generated by an amplicon and shotgun sequence simulator, Grinder [34], starting with a 2× coverage of each genome. The simulated metagenome was then fed to Xander with the eight Xander packages to assemble cas1. The assemblies were then subjected to chimera check by Xander. The HMM sensitivity was assessed by whether all the 17 subtypes of cas1 can be recovered from the simulated metagenome using the targeted-assembly method. The HMM specificity was evaluated by accurate mapping of the targeted-assembled nucleotide sequences to the cas1 coding region of the designated genome from the reference genome set using Bowtie2 [35]. The respective cas1 protein sequences were also searched against nr database (NCBI) by BLAST [36] to check the assembly accuracy.
Second, the curated reference genome set can better improve the performance of the eight HMMs within the Xander packages. We first confirmed that the cas1 sequences of the reference genomes were covered in the simulated metagenome by checking if at least two 45-mer of the reference cas1 was in the kmer set of simulated reads (Fig. 1b). For the reference cas1 that contributed to the simulated reads but was not captured by any of the preliminary models, we aligned the corresponding protein sequence of the reference cas1 to the eight HMMs and added it to the seeds and framebot.fa of the best aligned HMM to enhance the sensitivity and the recovery of cas1 diversity (Fig. 1b).
Similar to the simulated metagenomes, the preliminary assemblies of cas1 proteins from the Oklahoma soil metagenomes mentioned below were also used to improve the performance of HMMs for targeted assembly. We searched the preliminary cas1 assemblies (though short and below the length threshold) against nr database (NCBI) by BLAST [36] and added the near full-length best hits into the corresponding seeds and framebot files to optimize the models.
After the iteration by updating the coverage of the seeds and framebot files (Fig. 1a) and reconstructing the HMMs, the eight final Xander packages contain more robust models, which are available at https://github.com/Ruonan0101/Targeted_Cas1_assembly. The processing steps are summarized in Fig. 1a.
Sequence trimming and targeted cas1 assembly from Oklahoma soil metagenomes
The Illumina adapters were removed from the paired-end reads using Trimmomatic v0.33 [37] (ILUMINACLIP). The trimmed reads were filtered for contiguous segments longer than 45 bases (same as the kmer length used in assembling) with the average quality score higher than 20 (SolexaQA++ v3.1.5 [38] dynamictrim and lengthsort). The trimmed sequences were fed to Xander [10] for targeted cas1 assembly. All eight cas1 HMMs, together with bacterial and archaeal ribosomal protein L2 (rplB) HMMs, were run simultaneously on each sample from the same bloom filter with the same kmer size of 45 since this larger kmer size can result in better assemblies with lower chimera occurrence. The length cutoff for cas1 protein sequences was set to 60 amino acids based on the lowest length coverage of cas1 subtype, which was IIID at 30.8% (Table 1). We kept rplB sequences longer than 150 amino acids. RplB was used rather than the 16S rRNA gene because it is a single copy ribosomal protein in all microbes and is well tested for assembly by Xander and taxonomic placement [39].
Contigs validation and host assignment to phylum level
Because the eight HMMs have overlapping specificity, we first dereplicated the combined assemblies from all eight models before predicting host taxonomy. All contigs from one sample were pooled together and searched against NCBI nr database. The top five hits with the descending bit scores of each contig were recorded together with the information of percent identity, coverage, accession number, and lineage. The best hit was selected only if the percent identity of the top hit was greater than the second one by 3% or it has the same host taxonomy with the remaining hits (phyla level used in the following data analysis), otherwise the contig was unclassified. The unclassified contigs kept the information of the best hit but with lineage noted as “unclassified.” All the classified and unclassified contigs were sorted based on accession numbers of the best hit to check the potential overlap between models. Contigs assembled by different models but with the same accession number of the best hit were re-examined and the ones with (1) classified lineage, (2) higher bit score, and (3) longer length were selected. The host of cas1 assemblies was assigned based on the taxonomy annotation of the screened hits.
Validation of assembly accuracy by comparing to the traditional annotation method
We leveraged de-novo assembled contigs obtained from Oklahoma grassland metagenomes to validate the assembly accuracy and evaluate the performance of the targeted method. The quality-filtered metagenomic reads were assembled by MegaHit [40] using kmer from 31 to 131 with step size of 20. The option of -kmin-1pass was used to recover low coverage species. There were 1,488,684 contigs with length longer than 1000 bp used for the following analysis.
We then applied the traditional cas1 annotation method by searching the ORFs predicted from all the de-novo assembled contigs using the eight HMMs. The ORFs annotated as putative cas1 via hmmsearch (HMMER, v3.1b2, http://hmmer.org/) were further validated by NCBI nr database. As both targeted and de-novo assembly methods can generate gene fragments, we only compared the numbers of cas1 sequences with near full length (>300 aa) from both methods to make a fair and conservative estimate of the performance.
Validation of host assignment method by comparing to the cas1-mapped de-novo assemblies
The reliability of cas1-based host assignment was tested by searching the targeted-assembled cas1 sequences against the de-novo assembled contigs using BLASTn. A qualified match was defined as a hit with (1) the highest bit score, (2) the percent identity greater than 95%, and (3) the length coverage higher than 50%. We predicted the taxonomies of de-novo assembled contigs via the Contig Annotation Tool (CAT) [41] with modifications. CAT assigned a de-novo assembled contigs with NCBI lineage based on multiple open reading frames (ORFs) predicted by Prodigal [42]. As cas1 is normally grouped with other CRISPR-Cas genes, which are more prone to horizontal transfer [1], we removed ORFs with NCBI annotations as CRISPR-Cas related proteins and fed CAT with filtered ORFs for taxonomy assignment. To check the sequence redundancy of cas1-mapped de-novo assembled contigs, we used the single linkage clustering to cluster the de-novo assembled contigs [43]. The detailed clustering method and results were in Supplementary File 1.
cas1 subtype assignment and prevalence estimation
As cas1 protein sequences were mostly grouped by subtype, we assigned the subtypes by placing the assemblies on the cas1 protein tree. The tree was built with the 93 cas1 protein sequences of 17 subtypes from the reference genome set using FastTree [32]. The cas1 assemblies filtered from the validation step were aligned to a reference HMM and the modeled positions were used to map to the branches of the cas1 protein tree via pplacer [44]. The cas1 contigs then adopt the subtype assignment of the mapped references.
cas1 abundance was adjusted by subtracting the abundance of the contigs assembled from the potentially overlapped models. The adjusted cas1 abundance detected in a microbial phylum was further normalized by the corresponding rplB abundance and noted as cas1 prevalence of a particular phylum in the following discussion.
Statistical analysis
Sequence distance matrix was calculated using RDPTools (Clustering) and plotted in R (v3.3.3, Vegan, pheatmap [45]). Canonical correspondence analysis including the cas1 host composition with environmental attributes was conducted by R (v3.3.3) with packages of Vegan [46] and ggplot2 [47].
Source: Ecology - nature.com
