Patterns of R. irregularis transcriptome variation are congruent with patterns of R. irregularis genome variation
Phylogenetically diverse R. irregularis isolates significantly differed in their ability to colonize cassava roots, with colonization ranging from 21.7% of the root length colonized by SAMP7 to 72.9% by DAOM197198-CZ (Fig. 1c, Data S1). Colonization was significantly associated with the phylogeny of the fungus (Fig. 1c). Mock-inoculated control roots were confirmed to not be colonized by AMF. Despite the differences in fungal colonization, there were no significant differences in plant growth, measured as root and above ground dry weight (Fig. S1). This lack in trait differences is probably due to stage at which the plants were harvested, as cassava growth was shown to vary in response to different AMF but only at late stages of growth [16, 46]. The pattern of fungal gene transcription was clearly discernible among the fungal groups (Figs. 2a and S2A, B). The fungal phylogeny based on SNPs in the genome was congruent with a phylogeny based on transcript profiles (Fig. S2C). Approximately 5.9% of the gene repertoire (772 out of 13,109 genes) of the most divergent genetic groups (GP1 and GP4) were DT. These included important functions such as cell wall organization and biogenesis (Fig. 2b). These observations confirmed the strong divergence in genetic reprogramming between these fungal groups [21] during symbiosis.
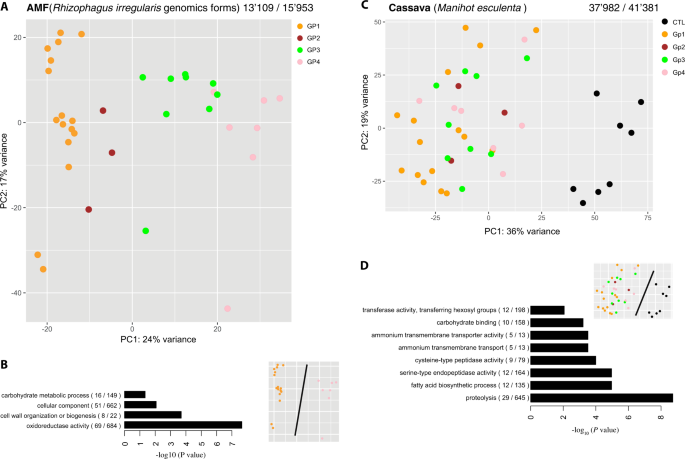
a PCA of fungal gene transcription of each R. irregularis isolate in cassava roots. The PCA, with mock-inoculated controls (CTL) treatment is shown in Fig. S2A. Colors represent the different genetic groups of R. irregularis. b GO enrichment bar plot for the predicted function of 772 differentially transcribed fungal genes between the two most divergent fungal groups (GP1 and GP4). c PCA of cassava root gene transcription and colored by treatment according to the fungal genetic group. d GO enrichment bar plot for the function of 353 genes that were differentially transcribed between all four fungal genetic group treatments and the CTL. The numbers above each PCA (a and c) represent the number of genes found as transcripts and the total number of annotated genes in the genome assembly of the species. Following each GO function shown in the bar plots (b and d), the number of genes is shown that were differentially transcribed compared with the total number of genes in that functional category. The small PCA representation above the bar plots (b and d) depicts the treatments compared to obtain the differentially transcribed genes displayed in the bar plots.
Upregulation of the fatty acid biosynthesis pathway in cassava in response to symbiosis
A comparison of gene transcripts from mock-inoculated control roots with transcripts of all R. irregularis inoculated roots revealed that the genetic reprogramming of the root, as a response to inoculation, affected less than 2.5% of the cassava gene repertoire (949 out of 37,982 genes). The transcription of only 353 cassava genes (0.93%) showed a conserved transcriptional response to symbiosis consistently across all R. irregularis isolates (Data S2a, b). Unlike fungal gene transcripts, patterns of cassava gene transcription did not cluster into identifiable groups based on the genetic background of the fungus, but were distinct from transcripts of the mock-inoculated plants (Fig. 2c). As a test of the robustness of the transcript dataset, we searched for orthologous genes of the so-called “mycorrhizal symbiosis toolkit” necessary for fungal colonization of roots. Of eleven orthologs, known to be necessary for mycorrhiza formation in Medicago truncatula, ten were significantly upregulated in cassava in the presence of mycorrhizal fungi (Fig. S3A, B). GO enrichment analysis revealed that differential transcription between mycorrhizal and non-mycorrhizal plants included genes involved in known essential functions of the symbiosis such as ammonium transport and carbohydrate binding (Fig. 2d). The fatty acid pathway (GO:0006633) was proportionally the most enriched and complete in the analysis. Some of these genes were also DT between plants inoculated with the most evolutionarily divergent isolates (GP1 and GP3 + 4). For example, this included one of the key enzymes of fatty acid synthesis, an enoyl-acyl carrier reductase (Manes.08G046300.1), a carboxyltransferase alpha (Manes.09G101700.2), and trans-2,3-enoyl-reductase (Manes.07G083100.1) (Data S2c).
We observed the upregulation of many cassava genes comprising the whole fatty acid pathway from initiation by transcription factors, through end of glycolysis, oxidative decarboxylation of pyruvate and the fatty acid synthesis pathway (Fig. 3, Data S4). Here we show for the first time that this pathway is commonly upregulated across all genetically different AM fungal treatments in an important global food security crop.
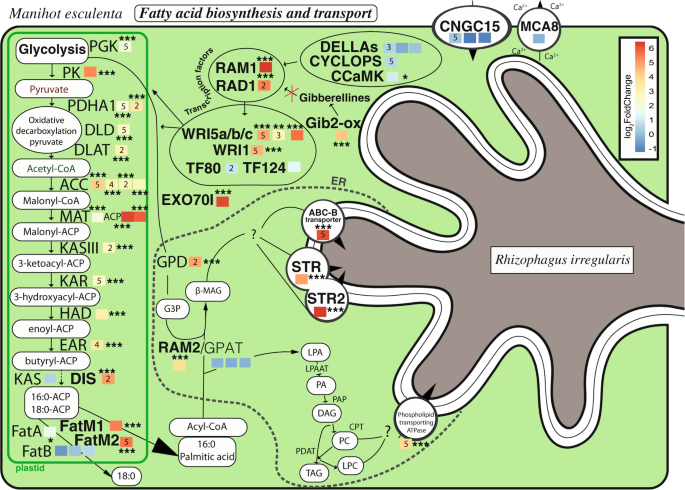
Genes commonly upregulated with all fungal isolates are represented in between their products. Colored squares next to each gene represent the log2fold change obtained with Kallisto and DESeq2. The asterisks represent the significance of the change (* < 0.05, ** < 0.01, *** < 0.001). Numbers within each colored square represent the number of indicators of a significant phylogenetic signal (out of five indicators) between the fungal phylogeny and the transcription of a given cassava gene. Genes that were shown to play a role, or putatively play a role, in fatty acid biosynthesis are represented. For a description of each gene acronym see Data S4.
Transcription factors
The transcription factor RAM1 (required for Arbuscular Mycorrhization 1) exhibited the largest fold change in transcription of all genes involved in fatty acid biosynthesis. It was proposed that RAM1 plays a central role in the activation of fatty acid biosynthesis [7]. Gibberellic acid inhibition induces the transcription of RAM1 [47]. Gibberellin-2-oxidase, that acts as a gibberellic acid inhibitor [48], was upregulated in mycorrhizal cassava (Fig. 3). This gibberellin-2-oxidase might, therefore, play an important role in the activation of RAM1 or other GRAS factors [49] (plant-specific proteins named after: GAI, RGA, and SCR). RAD1 (required for arbuscule development 1) is known to interact with RAM1 [50] and was shown to also be commonly strongly upregulated in cassava (Figs. 3 and S4A). WRI5a–c are transcription factors dependent on RAM1 [7]. Two WRI (WRI5a and c) orthologs were commonly upregulated (Figs. 3 and S4B). WRI5b upregulation was observed with one of the mapping strategies. In Arabidopsis thaliana these genes act at the end of the glycolysis and the mycorrhizal conserved genes WRI5 were confirmed as having a similar function in Nicotiana benthamiana [7, 13]. These findings confirm the tight link between these important and specific plant transcription factors with the process of fatty acid biosynthesis and their associated genes during AMF symbiosis.
Glycolysis and pyruvate oxidative decarboxylation
We observed common upregulation of genes involved at the end of the glycolysis and during the oxidative decarboxylation of pyruvate (Fig. 3) such as phosphoglycerate kinase (PGK) and pyruvate kinase, pyruvate dehydrogenase E1 component subunit alpha, dihydrolipoamide dehydrogenase (DLD), and dihydrolipoamide acetyltransferase component of pyruvate dehydrogenase. The genes involved in glycolysis are responsible for the production of pyruvate that will then be decarboxylated in the oxidative decarboxylation of pyruvate. At the end of this process acetyl-CoA, the precursor for fatty acid biosynthesis, will be released. In this study we showed, for the first time, that not only is the expression of the plant fatty acid biosynthesis pathway affected, but upstream plant molecular mechanisms are also impacted by AMF.
Fatty acid biosynthesis
Twelve genes of the fatty acid biosynthesis pathway were commonly upregulated (Fig. 3). An AM symbiosis-specific fatty acid synthesis pathway was commonly upregulated, including genes DIS (disorganized arbuscules, Fig. S4C) [15], FATM1 and 2 [51] (Fig. S4D), and RAM2 [52] (Fig. S4E). The other branch of this pathway (KASI, FATB, GPAT) was not (or poorly) upregulated. RAM2 is thought to produce β-monacylglycerol; a fatty acid exported to the outer membrane space [14]. RAM2 requires glycerol-3-phosphate. Glycerol-3-phosphate dehydrogenase gene was also commonly upregulated. Each gene and their variation for all treatments and their replicates are found in the heatmap in the supplementary information (Fig. S5A). The fatty acid biosynthesis pathway is, therefore, a commonly upregulated pathway in cassava during AMF symbiosis. As it has been suggested in model plants [7], this process is probably vital for the life of the fungus as well as probably essential for the symbiosis equilibrium in important tropical crops as well as model plant species.
A proposed transport route
Two ATP-binding cassette transporters (ABC-G), STR, and STR2, that were shown to be indispensable for arbuscule formation have been suggested as potential lipid transporters [14, 53]. Both were commonly highly upregulated in all fungal treatments (Fig. 3). Finally, we observed the common upregulation of a phospholipid-transport ATPase and an ABC-B transporter [54].
The common response across all fungal treatments of such a large number of genes involved in this fatty acid synthesis and export pathway in cassava, combined with the knowledge of those genes in other species and their conservation in mycorrhizal plants, indicates that the whole activation of this pathway is important for the symbiosis by producing more fatty acid as a currency of exchange with the fungi.
Fungal genetic variation and evolutionary history drive the cassava fatty acid pathway
Having established that the fatty acid pathway is indeed upregulated in cassava in response to all the genetically different fungi, we then asked whether the quantity of upregulation of each gene is associated with identifiable patterns of genetic variation in the fungus that represent their evolutionary history. We found that variation in transcription of a striking number of cassava genes in the initiation of the fatty acid pathway, fatty acid synthesis and transport was significantly associated with the pattern of genetic variation among R. irregularis isolates (Fig. 4a). This included 24 genes (Fig. 4a). Other commonly DT fatty acid genes (6) and suspected fatty acid-related genes (7) not presenting this pattern can be found in supplementary material (Fig. S5B) or in Data S2a. More specifically, there was a significant relationship between patterns of genetic variation based on 15229 genome-wide SNPs in the fungi and the amount of transcription of these genes (Fig. 4a). All genes showed the same pattern of response with higher transcription in response to GP1 and a lower induction of this pathway in response to fungi in GP3 and 4. AMF cannot synthesize their own fatty acids [8]. It is, therefore, intriguing that the high inducers of the plant fatty acid pathway were significantly the lowest colonizers and that the lowest inducers of the fatty acid pathway were significantly the highest colonizers (Figs. 1 and 4a).
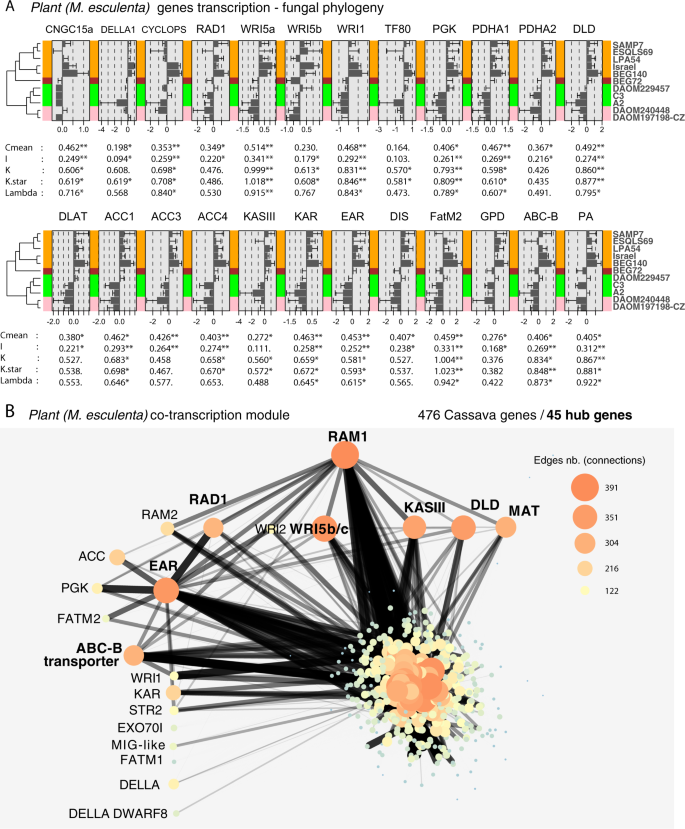
a Histograms showing the transcriptional responses of cassava genes in the different fungal treatments and represented as the difference of each treatment to the mean transcriptional response across all treatments. Bars represent ±S.E. Phylogenetic signal indicators and their significance are shown below in each histogram. Only genes that showed at least one significant indicator are displayed. Other fatty acid-related genes are shown in Fig. S5B. b The plant co-expression network based on one gene module comprising the largest number of genes from the fatty acid pathway. The network shows the strong co-transcription of RAM1 with several other transcription factors (WRI5c and RAD1) and fatty acid synthesis genes (DLD, KASIII, MAT, and EAR), which are also hub genes in this module (in bold). In this module, 45 were hub genes out of 476, but only the hub fatty acid-related genes are shown in bold. Edges (connections between nodes/genes) of the network are represented as a function of their weight, a measure of connection strength. Only the 5% strongest edges are represented with increase thickness and decrease transparency with connection intensity. Node size reflects the degree of connectivity, i.e., the number of connections with other nodes. More information on the network is available in Data S5a.
Gene network and hub genes of fatty acid biosynthesis revealed by genetic differences among R. irregularis isolates
We built co-transcription gene modules using the variation in transcription of all the M. esculenta genes in all the AMF inoculated treatments. Among the 31 cassava gene modules, one gene module contained most of the fatty acid-related genes. This module contained 476 genes, of which 45 were hub genes, i.e., hub genes have the highest connectivity within the module defined as a MM above 0.9 (MM > 0.9; Data S5a). RAM1 was the hub gene with the highest MM (MM = 0.97; Fig. 4b), i.e., it had the highest connectivity to all the other genes within the module. This reveals the gene hierarchy in the plant fatty acid synthesis pathway during AMF symbiosis, with RAM1 as one of the top initiators of this cascade. Other hub genes in the module were commonly upregulated in the pathway shown in Fig. 3, e.g., WRI5c (MM = 0.95), RAD1 (MM = 0.91), DLD (MM = 0.94), MAT (MM = 0.92), KASIII (MM = 0.93), EAR (MM = 0.96), and the ABC-B transporter (MM = 0.91). Other non-hub genes occurred in this module, including RAM2, FATM1 & FATM2, WRI5a & WRI1, EXO70I, STR2, MIG-like, KAR, ACC, and PGK. In this fatty acid module several functions were enriched such as carbohydrate metabolic process, carbon utilization, inorganic phosphate transmembrane transporter activity, and phosphate ion transport. Fatty acid functions were only marginally enriched in both the hub gene list (45 genes) and the module gene list (476 genes) without correction under the term FAS complex (P value = 0.00012, p-adjusted = 0.136) (Data S5b, c). This marginally significant enrichment might be due to the low-quality annotation, where several genes known to be implicated in the fatty acid pathway (RAM1, FATM1, 2, WRI5, and other genes) are not annotated as genes with a fatty acid function. Using the same methods, we combined cassava and fungal transcripts and built new symbiosis gene modules (27 modules). The module containing RAM1 (MM = 0.95), contained 165 hub genes (Data S5d) enriched in plant genes of the fatty acid biosynthesis process (p-adjusted = 0.000013) and FAS complex (p-adjusted = 0.01, Data S5e, f). However, no R. irregularis genes were hub genes in the module. While this might, at first glance, suggest a full control of the plant over the fungus, there are several reasons why this may not be the case. We caution such an interpretation from these results as they would be inconsistent with theory regarding the evolution of mutualistic symbioses, where mutualism is stable when neither partner can overexploit the other.
These findings show the central role played by RAM1 during AM symbiosis, and its co-transcription with a large number of well-known genes (WRI5, RAD1) confirm a strong link of this transcription factor with several key genes of fatty acid biosynthesis that were not previously been demonstrated, such as DLD, MAT, KASIII, and EAR.
Source: Ecology - nature.com

