Plasmids and strains
Our iSLC and SLC strains were both cultured in lysogeny broth (LB) media with 50 μg ml−1 kanamycin, 34 μg ml−1 chloramphenicol and 0.2% glucose for strains containing ColE1 origin and p15A origin plasmids in a 37 ∘C shaking incubator. Plasmid pTD103 RpaR-RpaI-LAA-sfGFP21, ptD103-CFP19, and pZA35-X174E(+LuxR)5 were constructed by our group in previous studies (Supplementary Fig. 7). Both plasmids pAM014 and pAM021 were obtained by inserting the gene 4CL2nt28 under the constitutive promoter J23106 from the Anderson promoter library. The 4CL2nt gene and the promoter were synthesized with overlapping PCR of long oligos (IDT). All plasmids were constructed by Gibson assembly followed by transformation into DH5α (Thermofisher) chemically competent E.coli. All plasmids were verified by Sanger sequencing before transformation into E.coli strain MG1655.
Microfluidics and microscopy
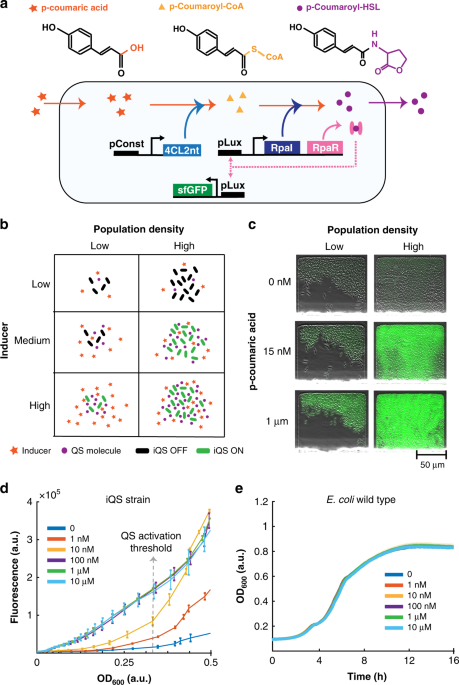
The microscopy and microfluidics techniques used in this study are similar to those previously reported by our group35. Our microfluidic devices were constructed from PDMS (poly-dimethylsiloxane), which was molded and baked on a silicon wafer with micron-scale features formed by cross-linked photoresist. Once the PDMS hardened, it was peeled off, and individual devices were cut out. In order to connect fluid lines to the device, holes where punched in correspondence of the inlets and outlets of the device. Afterwards, the devices were bonded onto glass slides using plasma activation. Before each experiment, the devices were left for 30 min in a vacuum chamber. Meanwhile, 1 ml of overnight cell culture was spun down by centrifugation and re-suspended in 10 μl of fresh media with appropriate antibiotics. After taking the device out of the vacuum chamber, a single droplet of re-suspended cells was positioned in correspondence of the outlet opening. Similarly, droplets of sterile fresh media were placed in correspondence of the inlets openings36. In all cases, 0.075% Tween20 was added to the medium to prevent cells from sticking to the PDMS walls. After all chip features were wetted, the fluids lines were plugged in and the height of the inlet was raised 10 to 40 cm above the device. The outlet syringe was instead placed at the same height of the device. For co-culturing experiments in Fig. 4, cells were cultured individually overnight and eventually spun down and re-suspended together (1:1 ratio) allowing for a single droplet to be loaded on the device. All experiments shown in Figs. 2 and 4 were performed in a side-trap array device with bacteria growth chambers ~100 × 80 μm in area and ~1.2 μm in height. The upstream channels consists of a series of dividing serpentine branches, which allow for sequential dilutions of the two input media, generating a gradient of eight different inducer concentrations (Supplementary Fig. 3). On the other hand, for the kill switch experiments, reported in Fig. 3, we used a simpler device with a single input and an ordered array of traps35. The dimensions of the traps are the same as the ones described for the gradient device. Experiments in Figs. 2 and 4 where carried out by connecting a syringe with LB + antibiotics + 0.075% Tween20 as inlet 1 and and a syringe with LB + antibiotics + 0.075% Tween20 + 1μM of p-coumaric acid as inlet 2. P-coumaric acid inductions for microfluidic experiments in Fig. 3b were performed by unplugging the syringe with pure media and substituting it with a second syringe containing media plus the appropriate acid concentration. For microscopy we used the same system as described in our previous work35. In brief, images were acquired with a Nikon TI2 using a Photometrics CoolSnap cooled charge-coupled device (CCD) camera. The scope and accessories were programmed using the Nikon Elements software. The microscope was housed in a plexiglass incubation chamber maintained at 37 ∘C by a heating unit. Phase-contrast images were taken at x4 and x10 magnification at 50–100 μs exposure times. At x4 magnification fluorescence exposure times were 2 s at 30% intensity for both gfp and cfp while at x10 magnification they were 200 μs at 30% intensity for both gfp and cfp. Images were taken every 6 min for each experiment. For induction experiments, imaging was paused while syringes were swapped.
Data analysis
Fluorescence intensity profiles were obtained by analyzing frames from the fluorescent channels. The mean fluorescence values were calculated by drawing a rectangle surrounding each trap individually and extracting the z-axis profile on ImageJ. Fluorescence values shown in Fig. 2 were normalized by dividing all data by a constant factor. In addition, the subset of traps for each column was normalized by subtracting the minimum value among the traps within the subset. Transmitted light data from Fig. 3b was normalized by subtracting the minimum value for each time trace. Fluorescence data shown in Fig. 4 was normalized by first dividing the subsets of traps by their overall maximum values and subsequently subtracting the respective minimum values. In addition, the data in Fig. 4 was smoothed with the command smoothdata() in Matlab. Heatmaps were generated in Matlab using the function heatmap(). For the co-culture experiments, when overlap between the gfp and cfp channels was observed, values were corrected taking into consideration the image frames in order to subtract overlapping signal.
Plate-reader experiments
For plate-reader experiments, the appropriate strains were seeded from a −80 ∘C glycerol stock into 3 ml LB with 0.2% glucose and appropriate antibiotics and incubated in a 37 ∘C shaking incubator. The following day, 2 μl of overnight culture were added to 200 μl of fresh media with appropriate antibiotics in a standard Falcon tissue culture 96-well flat bottom plate. Cells were incubated at 37 ∘C shaking in a Tecan Infinite M200 Pro. Cells were grown for about 12 h. The OD at 600 nm absorbance was measured every 10 min.
Cells survival assay
Cell viability assay to test the efficacy of iQS as a kill switch was done measuring colony forming units (CFUs), following a protocol found in the literature37. Cells were grown under survival conditions in LB with 0.2% glucose, which inhibits the LuxI promoter thanks to the presence of a binding site for the CAP-cAMP activating complex38. In the morning, they were transferred into four liquid cultures of fresh LB medium with 0.2% glucose, 300 nM p-coumaric acid, 500 nM p-coumaric acid and 1 μM p-coumaric acid, respectively. Samples were collected every 2 h and serially diluted in PBS over a 7-log range and spotted (2 μl) onto LB agar plates with 0.2% glucose. The equations used are: CFU/ml = (number of colonies) × (dilution factor)/0.002 mL, survival ratio (log10) = log(CFU/ml with glucose)/(CFU/ml with p-coumaric acid).
Modeling
We constructed a deterministic model to qualitatively describe the dynamic behavior of the iSLC strain. The model is based on a set of six ordinary differential equations (ODEs), which track the evolution of the following six variables: the cell number into a single microfluidic trap (N), the external concentration of p-coumaroyl-HSL (pH), the intracellular concentration of lysis protein (L), the intracellular concentration of the pC-HSL synthase RpaI (I), the intracellular concentration of p-coumaric acid-CoA ligase (E) and the intracellular concentration of the intermediate compound p-coumaroyl-CoA (pA). The inducer concentration is defined as a fixed parameter.
A non-zero p-coumaric acid concentration induces the production of the intermediate molecule p-coumaroyl-CoA (pA) through the conversion mediated by the p-coumaric acid-CoA ligase (encoded by gene 4CL2nt) (E). The production term of the latter is defined as a constant variable according to the constitutive promoter, which drives it. The intermediate product (pA) is eventually transformed into the quorum sensing molecule pC-HSL through the RpaI synthase enzyme (I). Once a threshold value of extracellular pC-HSL is reached, the intracellular production of the pLux promoter driven genes (RpaI, RpaR, and E) are brought to the ON state. The same promoter also drives the lysis gene, therefore positive feedback also results in cell lysis. We assume that the quorum sensing molecule diffuses quickly through the membrane, therefore we do not distinguish between intracellular and extracellular HSL concentration. In addition, we assume that the pC-HSL-RpaR complex binding is instantaneous, so that the model can be simplified by ignoring the dynamics of the binding complex. Degradation of all proteins (L, I, E) is associated with dilution due to cell growth (μG), as well as basal intracellular degradation (γL and γI). In addition to those terms, RpaI (I) is also actively degraded by ClpXP proteases (γC). Overall, our model can accurately predict the three main dynamics of the bacterial population as the inducer concentration is varied. With zero inducer concentration, the population grows reaching a steady state value. Similarly, at very small inducer concentrations, the population undergoes small amplitude lysis events followed by steady state. We observed a finite range of intermediate inducer values, which resulted in sustained oscillations of population density. Finally, we observed total population death with no survivors for high concentrations (Supplementary Fig. 8). We can visualize the non-linear dynamics of this system using phase portraits obtained by plotting N (cell number) against L (lysis protein) (Supplementary Fig. 9). As the p-coumaric acid concentration is increased, the simulations show a first transition from a stable spiral to a limit cycle, which indicates sustained oscillations. A further increase in the inducer parameter causes the limit cycle to disappear in favor of a stable fixed point. All plots were generated in MATLAB.
$$frac{{mathrm{d}}N}{{mathrm{d}}t}={mu }_{G}* N* ({N}_{0}-N)-N* frac{k* {L}^{n}}{{({L}_{0})}^{n}+{L}^{n}}$$
(1)
$$frac{{mathrm{d}}pA}{{mathrm{d}}t}={mu }_{4}* {mathrm{inducer}}* E-{gamma }_{CoA}* pA-{mu }_{G}* pA$$
(2)
$$frac{{mathrm{d}}pH}{{mathrm{d}}t}={mu }_{H}* N* I* pA-frac{u* pH}{1+frac{N}{{N}_{0}}}$$
(3)
$$frac{{mathrm{d}}L}{{mathrm{d}}t}={C}_{l}* left({alpha }_{0}+frac{{alpha }_{H}* {left(frac{pH}{{H}_{0}}right)}^{4}}{1+{left(frac{pH}{{H}_{0}}right)}^{4}}right)-{gamma }_{L}* L-{mu }_{G}* L$$
(4)
$$frac{{mathrm{d}}I}{{mathrm{d}}t}={C}_{i}* left({alpha }_{0}+frac{{alpha }_{H}* {left(frac{pH}{{H}_{0}}right)}^{4}}{1+{left(frac{pH}{{H}_{0}}right)}^{4}}right)-{gamma }_{I}* I-{mu }_{G}* I$$
(5)
$$frac{{mathrm{d}}E}{{mathrm{d}}t}={C}_{l}* {P}_{{mathrm{const}}}-{gamma }_{4}* E-{mu }_{G}* E-{gamma }_{C}* E$$
(6)
We chose model parameters based on a similar model previously published5. Compared to the Lux quorum sensing system, the iSLC showed higher promoter leakiness, which was taken into account by increasing the basal production term (α0). The parameter values used in the model are μG = 0.2 (dilution due to cell growth), N0 = 20 (cell capacity of a single trap), k = 10 (maximum rate of cell lysis), L0 = 1 (concentration of lysis protein resulting in half maximum lysis), n = 2 (Hill’s coefficient), Cl = 0.5 (copy number of the lysis gene), Ci = 1 (RpaI gene copy number), Pconst = 20 (strength constitutive promoter driving gene 4CL2nt), γ4 = 2 (degradation of enzyme p-coumaric acid-CoA ligase), α0 = 1 (pLux basal leakiness), αH = 20 (Lux promoter pC-HSL-RpaR induced production), H0 = 4.5 (pC-HSL-RpaR binding affinity to pLux), γL = 2 (lysis protein basal degradation), γI = 2 (RpaI protein basal degradation), γC = 12 (RpaI protein degradation due to ClpXP), γCoA = 2 (p-coumaroyl-CoA basal degradation), μ4 = 30 (conversion rate of p-coumaric acid into p-coumaroyl-CoA), μH = 15 (pC-HSL production rate) and μ = 12 (maximum AHL clearance rate due to flow).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Source: Ecology - nature.com
