Ethics statement
The sugarcane borer T. intacta larvae and adult moths were reared on artificial diet in the laboratory of Guangzhou Sugarcane Industry Research Institute (23°8′N, 113°17′E, Guangzhou City, Guangdong Province, China). A laboratory population was kept and maintained at 27 ± 1 °C, 70 ± 10% RH, and 14:10 h L:D. The host-plant sugarcanes (ROC22) were cultivated in the experimental field of Guangzhou Sugarcane Industry Research Institute. All experimental animal procedures including this pest were approved by the Institutional Review Board at Central China Normal University in China (CCNUIRB).
Field experiment
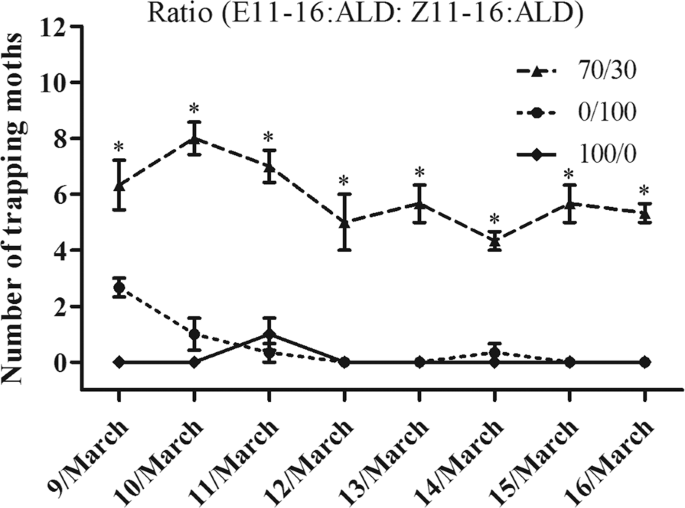
In the field trapping experiment, synthetic pheromone-baited lures will trap and capture male moths inside the experimental device. Proper trap design was critical to kill the pest once it enters the trap. Different ratios of sex pheromone components (E11-16Ald: Z11-16Ald: 0/100, 100/0 and 70/30) were used to attract T. intacta in the field trials in Guangdong Province, China (20.7°N, 110.2°E). Water basin traps were used to capture sugarcane borers, which consisted of a basin and a lure hooked together by an iron wire. The iron wire passed through the basin by two holes with a lure 2 cm of the top of the water, which every trap was 50 meters apart. The traps were checked at 6 am every day and recorded the captures of T. intacta.
In addition to the traditional rubber bait, we also used a new matrix material in the trapping test, which consisting of more polymeric compound with a melting point below 100 °C. The polymer/pheromone combination was emulsified and dispersed in water. And the sex pheromones were dissolved in the matrix material together with an antioxidant. This mixture was then emulsified using a surfactant at a temperature above 80 °C. After then, these new baits were used to measure their trapping effect in the sugarcane field. p < 0.05 indicated significant difference by Student t-test.
Cloning and sequence analysis of olfactory gene from T. intacta
The TintPBP4 gene was amplified from T. intacta by PCR using primers, which were designed according to the gene sequence in the sequencing results of the transcriptome. PCR was performed: a denaturation step at 94 °C for 3 min; and followed by 30 cycles of 94 °C for 30 s, 56 °C for 30 s and 72 °C for 45 s and final extension for 5 min at 72 °C. PCRs were separated on a 1.0% agarose gel. Products in the estimated size range were excised, purified with a TaKaRa MiniBEST Agarose Gel DNA Extraction Kit Ver.4.0 (TaKaRa) and subcloned into pEASY-T1 vector, and positive clones were sequenced. The correct pEASY-T1-PBP plasmid was extracted as PCR template and amplified by gene specific primers with protective bases and restriction sites (BamHI and HindIII).
The TintPBP4 cDNA sequence was analyzed using the online program BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) and signal peptide was predicted by Signal P 4.1 Server (http://www.cbs.dtu.dk/services/SignalP/). The amino acid sequences multiple alignments of TintPBP4 and other PBPs from Lepidopteran insects were analyzed and aligned using the DNAMAN software. The theoretical isoelectric point of TintPBP4 protein was obtained through the website (http://web.expasy.org/cgi-bin/protparam/protparam). The phylogenetic tree analysis of TintPBP4 with similar PBPs of other insect species were constructed by MEGA 6 software.
Quantification of relative tissue expression
Quantitative real-time PCR test was determined by a Bio-Rad CFX 96 real-time PCR system with SYBR Green I fluorescent dye. Olfactory organ antennae and different tissues including heads (without antennae), thoraxes, abdomens, legs and wings were collected and total RNA was extracted using Total RNA kit I. cDNA was synthetized from 1 μg of total RNA using Reverse Transcriptase kit according to the manufacturer’s protocol. The mRNA transcript of TinfPBP41 was assessed by QRT-PCR with the specific primers (Table 1). The QRT-PCR reaction was performed in a final volume of 20 μl reactions containing 2 μl of cDNA (diluted 5 times from original cDNA concentration), 0.4 μl of each primer, 10 μl of TransStart Tip Green QPCR SuperMix (TransGen), and 7.2 μl of RNase-free water. The cycling conditions were as follow: 95 °C for 3 min; 40 cycles at 95 °C for 10 s and at 50 °C for 30 s; and melt curve at 65 °C to 95 °C for 5 s. Each tests included three biological replicates and three technical repetitions. The relative expression levels were calculated by the 2−△Ct method, and SigmaPlot 10.0 was used to draw the histogram. p < 0.05 indicated significant difference by Student t-test.
Recombinant expression of olfactory protein
The 7.5 μl purified PCR product was ligated into cloned into 1 ul the pET-32a (+) vector by incubating the mixture with 0.5 μl T4-DNA ligase and 1 μl 10 X T4 ligase buffer at 4 °C for 16 h. The recombinant plasmid pET-32a-TintPBP4 was transformed into E. coli DH5α competent cells, positive colonies were selected by their ampicillin resistance. And the constructed recombinant expression vectors were further confirmed by sequencing. Subsequently, the plasmid was transferred into E. coli BL21 (DE3) competent cells, and the target proteins were expressed according to a previously reported operating procedures. The crude protein fractions were further purified from the supernatant using an affinity chromatography column. The purified protein fractions were analyzed by 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and stored in −80 °C until use.
Fluorescence binding assay
The binding assays were conducted following our previous studies10. First, we tested the binding of a fluorescent probe N-Phenyl-1-naphthylamine (1-NPN) to the protein. Next, the ligand compounds were measured in fluorescence competitive binding assays using 1-NPN as the fluorescent reporter (0.5 μM), and 0.5–10.0 μM for each competitor. Two sex pheromones from T. intacta were carried out in binding assays. Dissociation constants of bound ligand were calculated from the corresponding half maximal inhibitory concentration (IC50) values, using the equation: KD = [IC50]/(1 + [1-NPN]/K1-NPN), where 1-NPN is the free concentration of 1-NPN and K1-NPN is the dissociation constant of the Protein/1-NPN complex.
Source: Ecology - nature.com
