Sampling
Six species were selected, which are characteristic, widespread, abundant, and often co-occurring elements of the Eurasian steppes. In addition, heteroploid, taxonomically difficult species were avoided, the exception being A. onobrychis. Samples of the plant species A. onobrychis L. (Fabaceae), E. seguieriana Neck. (Euphorbiaceae), and S. capillata L. (Poaceae), and the animal species P. taurica Santschi, 1920 (Formicidae), O. petraeus (Brisout de Barneville, 1856), and S. nigromaculatus (Herrich-Schäffer, 1840) (both Acrididae) were collected between 2014 and 2017. All species were sampled across their distribution ranges, while extrazonal occurrences were more densely sampled than zonal ones. Of the six study species, only S. capillata was suggested to be widespread in the Iberian Peninsula. There, S. capillata was not exhaustively sampled outside the Pyrenees, as preliminary data suggested that the Iberian populations belong to another, cryptic species. This divergence is supported by the classification of the Iberian steppes as Mediterranean, instead of Central Asian, vegetation type35. In total, 456 populations from 320 localities were sampled (details are given in Supplementary Data 1). Identification of animals at the species level was done using the corresponding keys (grasshoppers36, ants37). Collected specimens were stored in silica gel (plants) or 96% ethanol (animals) for further analyses; herbarium vouchers and animal specimens are stored at the Departments of Botany (herbarium IB) and of Ecology, University of Innsbruck, respectively.
DNA extraction and RADseq library preparation
Plant DNA was extracted from leaf tissue with a sorbitol/high-salt cetyltrimethylammonium bromide method38 and purified using the NucleoSpin gDNA clean-up kit (Macherey-Nagel, Düren, Germany). Animal DNA was extracted from leg muscle tissue (O. petraeus and S. nigromaculatus) or whole animals (P. taurica) with the DNeasy Blood & Tissue Kit (Qiagen, Düsseldorf, Germany). Altogether, 370 populations from 266 localities were sequenced by RADseq (for details see Supplementary Table 1, Supplementary Table 6). Prior to the RADseq analyses, the genome size of each species was determined with flow cytometry to aid the selection of suitable restriction enzymes39. RADseq libraries were prepared using published protocols with minor modifications40 (for details see Supplementary Material).
Identification of RADseq loci and SNP calling
The raw reads were quality filtered and demultiplexed according to individual barcodes using Picard BamIndexDecoder (included in the Picard Illumina2bam package; available from https://github.com/wtsi-npg/illumina2bam) and the program process_radtags.pl implemented in Stacks41. All resulting raw RADseq data are available in the NCBI Short Read Archive (accession numbers in Supplementary Data 1).
Before calling SNPs from the demultiplexed Illumina reads, the data were species-specifically preprocessed. Large genomes, as observed in O. petraeus and S. nigromaculatus, are known to contain large portions of pseudogenes, transposable elements, and noncoding DNA42. These elements are prone to cause problems in SNP calling procedures, as homology of a fragment cannot be ensured if multiple copies are present in an organism’s genome. To address this issue, reads of O. petraeus and S. nigromaculatus were mapped to a reference genome. To do so, RepeatMasker (A.F.A. Smit, R. Hubley & P. Green RepeatMasker at http://repeatmasker.org) was used to identify and mask repeated elements present in the Locusta migratoria genome42 (GenBank: AVCP000000000.1). In the next step, the quality-filtered RADseq reads were mapped to the masked L. migratoria genome using Stampy v. 1.0.2043. Mapped reads were selected with Samtools44, and only those were further used in downstream analyses, starting with ref_map.pl of Stacks41.
Apart from the two grasshopper taxa, RAD loci were assembled, and single nucleotide polymorphisms (SNPs) were called de novo, using the denovo_map.pl pipeline in Stacks version 1.4641. Settings for the denovo_map.pl program [i.e. a maximum number of differences between two stacks in a locus in each sample (−M), and a maximum number of differences among loci to be considered as orthologous across multiple samples (−n)] were separately evaluated and optimised for each species, considering the number of potentially paralogous loci based on trial analyses (Supplementary Table 1).
The function export_sql.pl in the Stacks package41 was used to extract loci information from the catalogue. RAD tags were removed if (i) in any of the samples more than two alleles were detected, to reduce the risk of including paralogues in the dataset; (ii) the number of deleveraged tags was higher than 0. The programme populations implemented in the software Stacks41 was used to export the selected loci, whereas whitelists were used to exclude the unwanted loci as described above. For the first dataset, later used for Bayesian clustering, a set of RAD loci was exported into STRUCTURE45 format using the –structure and –write_random_snp flags41. The latter command was used to select only a single, random SNP per fragment to minimise the chance of selecting linked loci for the Bayesian clustering. For this dataset, all individuals sampled from the same site were defined as a population. For a second dataset per species, later used for phylogenetic tree reconstruction, concatenated RAD loci were exported as sequence alignment in phylip format using the –phylip_var_all flag in Stacks41.
Genetic clustering and phylogenetic reconstruction based on RADseq data
The optimal grouping of the populations was calculated using Bayesian clustering in STRUCTURE (version 2.3.4), using the admixture model with uncorrelated allele frequencies45. The analyses were performed separately for each species with input data characteristics specified in Supplementary Table 1 and the number of analysed individuals and populations shown in Supplementary Table 6. Ten replicate runs for K (number of groups) ranging from 1 to 10 were carried out using a burn-in of 200,000 iterations followed by 2,000,000 additional MCMC iterations. The optimal number of groups (K) was identified by identifying where the increase in likelihood started to flatten out, the results of replicate runs were similar, and the clusters were non-empty. Additionally, the deltaK criterion was employed, reflecting an abrupt change in likelihood of runs at different K46.
The concatenated RAD-locus alignments of each species were analysed as follows: Phylip alignments were imported into R (version 3.4.4) using the R package phrynomics (https://github.com/bbanbury/phrynomics.git). Subsequently, loci and individuals that contained more than 75% missing or ambiguous base calls at polymorphic sites (more than 85% in O. petraeus) were removed for downstream phylogenetic analyses (Supplementary Table 1). This step was done to meet the criteria proposed for robust phylogenetic inference from SNP data47. To infer phylogenetic relationships, maximum likelihood (ML) phylogenies were calculated using RAxML (version 8.2.8)48. All tree searches were done under a general time reversible model (GTR) with categorical optimisation of substitution rates (ASC_GTRCAT)48. Optimal substitution models were selected beforehand via the smart model-selection algorithm49. The best-scoring ML trees were bootstrapped using the frequency-based stopping criterion50. All phylogenies were initially rooted using an outgroup (Supplementary Table 7, not shown in the final figures). As the final alignments used for phylogenetic inference contained only variant sites, Felsenstein’s ascertainment bias correction implemented in RaxML was used to account for the missing invariant sites as recommended51. In addition to these complete phylogenies, in which all accessions passing the defined criteria were used, an additional population phylogeny, containing only one random individual per sampled population, was calculated for each species, following the same methodology. These population trees were later used to display the relationships between sampled populations (Fig. 3). The number of populations and individuals used to infer both phylogenies is given in Supplementary Data 1 and Supplementary Table 6.
Ecological niche modelling and paleo-projections
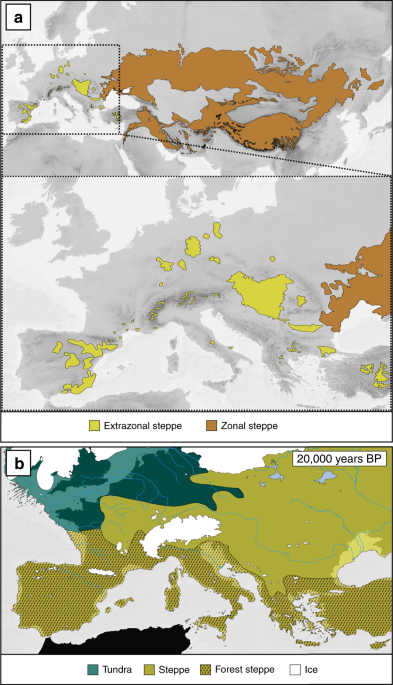
To avoid background selection and sampling bias, sparsely sampled Eurasia east of the Crimea was excluded for ENM52,53,54. Model overfitting due to spatial autocorrelation55 was reduced by removing localities within a spherical distance of 5 km using PASSaGE2 v. 2.0.11.656. The final dataset comprised 380 records (details in Supplementary Data 1, Supplementary Table 6). Bioclim variables for current climatic conditions were obtained from Worldclim v.1.4 (http://www.worldclim.org/)57 at a resolution of 30 arc-seconds and clipped to the area of study encompassing the European distribution of the six steppe species. To compensate Worldclim’s low precision for precipitation variables in mountainous areas57, observational data of 23 alpine climate stations were used to generate interpolated precipitation layers for the Alps (see Supplementary Methods, Supplementary Fig. 6, Supplementary Table 8). Pairwise correlation between variables was assessed using ENMTools (version 1.4.4)58, and variables with a Pearson’s correlation coefficient > 0.9 were removed based on expert knowledge. Two variables (bio18, bio19) were excluded for technical reasons (Supplementary Methods). For the final models, eleven variables were used (bio2, bio3, bio4, bio8, bio9, bio10, bio11, bio12, bio15, bio16, bio17; see Supplementary Methods). All ENMs were generated using MAXENT (version 3.3.3.k)59. Model parameters and details on the replication method are described in Supplementary Methods.
Species-specific tuning and jackknife tests (for details, see Supplementary Methods, Supplementary Table 3) were done to improve model performance and transferability60. The resulting models were projected to conditions of the Last Glacial Maximum, based on MIROC3.221 and CCSM321, at 30 arc-seconds spatial resolution. The projected variables were restricted to values encountered in model calibration under current conditions using clamping. To quantitatively assess projection uncertainty, MESS were used, whereby negative and positive MESS scores indicate non-analogue and analogue climates, respectively20. To reduce uncertainty stemming from different models, consensus prediction maps were generated by averaging over both paleo-projections (CCSM and MIROC)61. Areas of Stability were defined as grid cells that received higher suitability than the Maximum Training Sensitivity Plus Specificity (MTSS) threshold and were not covered by ice during Pleistocene cold stages62. Average Suitability of Stable Area maps were calculated as mean suitability of the individual consensus predictions after the MTSS had been applied. Projections and MESS analyses of all study species are presented in Supplementary Fig. 5.
PD and spatial patterns of diversity
To evaluate how much of each species’ diversity was represented by its extrazonal distribution compared with its zonal distribution, PD was calculated for the respective area17. To account for uneven sampling between the zonal and extrazonal steppes, the rarefaction approach implemented in the R package phylorare was used63. For these calculations, the complete maximum likelihood phylogenies were used (see section above).
PD complementarity was put in a spatial context to assess the uniqueness and heterogeneity of the extrazonal steppes. To do so, geographic entities, that is, grid cells of one-degree edge length (i.e. approximately 100 km), were defined for the extrazonal steppes prior to the analysis. For each of these grid cells, PD complementarity was calculated (i.e. the branch length represented within the grid compared with a reference set of branches)17. This was done based on the complete ML phylogenies (see section above), using all branches occurring in the zonal steppes as a reference. Because of the sampling-related unequal number of branches per grid cell, a random downsampling approach was implemented. In this stepwise procedure, the full dataset of each taxon was randomly reduced in such a way that the downsampled dataset still contained all grid cells but only one single branch per grid cell. For each of these reduced data sets, PD complementarity was calculated using all zonal branches as reference. After repeating this step 100 times per taxon, median PD complementarity values were calculated for each grid cell. To enable comparability across taxa, these values were transformed via division by the respective maximum median value obtained for a particular taxon so that the final values ranged between 0 and 1. Downsampling was done via custom R scripts, and the calculation of all PD complementarity was done in PDA (version 1.0.3)64. Additionally, PD complementarity for both all extrazonal steppes and all zonal steppes was calculated for each taxon. This was done to compare the amount of unique PD within each area, again using PDA (version 1.0.3)64.
Mitochondrial DNA sequencing
For two species, O. petraeus and P. taurica, parts of the mitochondrial COI gene were sequenced. Mitochondrial DNA sequencing in grasshoppers is prone to pseudogene amplification65. Hence, species-specific primers were designed using a cDNA template of the COI gene. To do so, total RNA was extracted from O. petraeus hind femur tissue, which had been stored at −80 °C immediately after field collection, using the Qiagen RNeasy Micro Kit (Qiagen, Düsseldorf, Germany). Extracted RNA was transcribed into cDNA via RevertAid reverse transcriptase (Thermo Fisher Scientific) in a 60-minute incubation step at 42 °C followed by an enzyme-deactivation step at 70 °C for 10 minutes. Using the cDNA as a template, the COI gene was amplified using published primers under standard PCR conditions (Supplementary Table 2). The resulting fragment was used to design specific primers inwards to the original binding sites. Primer quality and specificity were evaluated in silico using fastpcr (version 6.0)66. Specific primers were then used to amplify the COI gene from the DNA extracts previously prepared for RADseq using the Rotor-Gene Probe PCR Master mix (Qiagen, Düsseldorf, Germany) (Supplementary Table 2). For amplification of COI of P. taurica, published standard primers were used (Supplementary Table 2). Oligonucleotide synthesising and all Sanger sequencing steps were done by Eurofins Genomics, Ebersberg, Germany. All generated sequences were uploaded to NCBI GenBank (accession numbers in Supplementary Data 1).
Molecular dating of divergence based on mitochondrial DNA sequences
A mitochondrial mutation rate of 0.01615 substitutions per site per million years (My), which equals 3.2% divergence per My, previously proposed for European arthropods67, was used to date divergence events in the COI-based phylogenies of P. taurica and O. petraeus. A Bayesian uncorrelated lognormal relaxed clock as implemented in BEAST v.1.868 was applied to estimate the branch lengths. Before, the best substitution model, TrN+G for P. taurica and GTR + I + G for O. petraeus, was identified with jModelTest v.2.1.469 by using the corrected Akaike Information Criterion. The clock rate was modelled with a lognormal distribution and a mean of 0.016 substitutions per site per My, a standard deviation of 0.2 and an offset at 0.067. Two independent analyses were run for a total of 10,000,000 generations. Log files were analysed using TRACER v.1.570 to assess convergence and to ensure that the effective sample size (ESS) for all parameters was >200. The resulting trees were combined using LogCombiner v.1.7.568 with a burn-in of 25%. Subsequently, a maximum clade credibility tree was constructed using TreeAnnotator v.1.7.570.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Source: Ecology - nature.com
