DNA extraction and stLFR library construction, and sequencing
The long genomic DNA molecules were extracted from the muscle of Mekong tiger perch using a conventional method for sufficient DNA quality31. The stLFR library was constructed following the standard protocol using MGIEasy stLFR library preparation kit (PN:1000005622)12. In detail, the transposons with hybridization sequences were inserted in the long DNA molecules approximately every 200–1000 base pairs. The transposon integrated DNAs were then mixed with beads that each contained an adapter sequence. A unique barcode was shared by all adapters on each bead with a PCR primer site and a capture sequence that was complementary to the sequences on the integrated transposons. When the genomic DNA was captured to the beads, the transposons were ligated to the barcode adapters. After a few additional library-processing steps, the co-barcoded sub-fragments were sequenced on the BGISEQ-500 sequencer. To generate long reads to overcome the gaps (long ambiguous sequences) induced by repeats, library preparation and sequencing were performed on the MinION nanopore sequencer (Oxford Nanopore Technologies, Oxford, UK) according to the base protocols from Oxford Nanopore.
The adult individual from the Mekong river was purchased from the farmers’ market in Lao People’s Democratic Republic on May 21, 2018, and sex was unknown. The experimental procedures were in accordance with the guidelines approved by the institutional review board on bioethics and biosafety of BGI (IRB-BGI). The experiment was authorized by IRB-BGI (under NO. FT17007), and the review procedures in IRB-BGI meet good clinical practice (GCP) principles.
Reads filtering, genome size estimation, and genome assembly
We generated a total of 1,223,801,322 million raw pair-end co-barcoding reads of 122.4 Gb. To obtain a high-quality genome, SOAPnuke (v2.2)32 was performed to filter low-quality reads (>40% low-quality bases, Q < 7), PCR duplications, or adapter contaminations. After that, 753,357,182 clean pair-end reads remained. Based on the 17-mer analysis, the Mekong tiger perch genome size was estimated to be 623 Mb. Supernova assembler v2.0.1 (10X Genomics, Pleasanton, CA) was used to construct contigs and scaffolds, followed by gap closing using GapCloser (v1.2)14. We generated a total of 11.0 Gb long reads on the MinION nanopore sequencer and further filled the gaps using TGSGapFiller15 with default parameters.
Repeats prediction, gene structure prediction, and gene function annotation
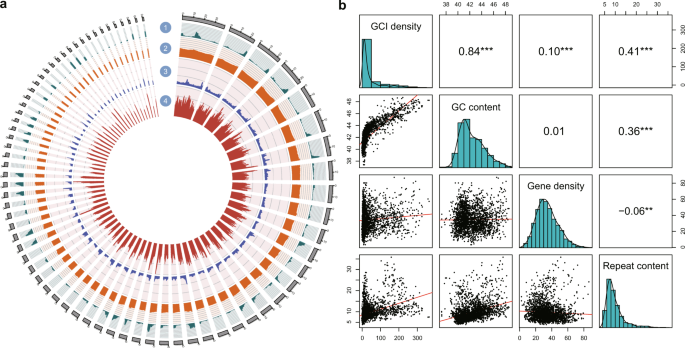
To predict repeat elements in the Mekong tiger perch genome, we used both de novo approaches and homology-based approaches. Firstly, we aligned our genome against the Repbase database33 at both protein and DNA levels by using RepeatMasker (v4.0.5) and RepeatProteinMasker (v4.0.5)34 to identify transcriptional elements (TEs). Secondly, we used RepeatModeler (v1.0.8)35 and LTR-FINDER (v1.0.6)36 to implement de novo repeat annotation. Next, we used RepeatMasker to complete repeat elements identification and classification. Lastly, we combined the above results.
We masked the repeats in Mekong tiger perch genome and gene prediction was performed using both homology-based and ab initio prediction. For homology-based annotation, we downloaded protein sequences of Dicentrarchus labrax, Labrus bergylta, Larimichthys crocea, and Gasterosteus aculeatus from NCBI. We aligned these sequence to Mekong tiger perch genome using BLAST37 with an E-value cutoff of 1e−5 and coverage >30% to identify homologous genes. Based on the aligned results, we used GeneWise (v2.4.1)38 to predict gene models. Furthermore, we used AUGUSTUS (v3.1)39 and GENSCAN (v2009)40 for ab initio prediction with default parameters and zebrafish data as a training set. Lastly, we integrated all above gene models by EVM41. We used BUSCO (v3.0.2)42 to assess gene annotation integrity using metazoan (v9) database.
To perform gene function annotation, we aligned the predicted gene sets against Kyoto Encyclopedia of Genes and Genome (KEGG, v87.0)43 and NR (v84)44 databases using BLASTP37 to identify genes with similar functions (E-value ≤ 1e-5). For identifying gene motifs and domains and obtaining Gene ontology (GO) terms45, we aligned our predicted genes against ProDom46, Pfam47, SMART48, PANTHER49, and PROSITE50 using InterProScan51.
Prediction of ncRNA and CpG islands
Four types of ncRNA (Non-coding RNA), including tRNA, snRNA, miRNA, and rRNA were predicted. We used tRNAscan-SE (v1.3.1) to predict tRNA in our genome with default parameters. The genome was aligned against Rfam (v12.0) database (Nawrocki E P et al., 2015) and based on mapping results we used infernal (v1.1.1) (Nawrocki E P & Eddy S R, 2013) to infer snRNA and miRNA. We aligned vertebrate rRNA database against Mekong tiger perch genome to predict rRNA.
The CpG islands (CGIs), which are clusters of CpGs in CG-rich regions, were identified on genome wide using CpGIScan52 with the parameters “–length 500–gcc 55–oe 0.65”.
Comparative genome analysis
We downloaded the annotation files of eight species including Dicentrarchus labrax, Gasterosteus aculeatus, Labrus bergylta, Labrus bergylta, Notothenia coriiceps, Oplegnathus fasciatus, Larimichthys crocea, Takifugu rubripes, and Sparus aurata form NCBI database (Supplementary Table S7). The longest transcript was extracted for each gene. We filtered out the sequences with length less than 50 amino acids, termination codon in the middle, and the sequence length not divisible by 3 to obtain high-quality gene sets for each species.
All-versus-all protein similarities were precomputed using BLASTP37 and TreeFam (v4.0)53 was used to identify gene families. We concatenated single-copy genes into a supergene matrix for all species and extracted sites on the first phase of codon to construct the phylogenetic tree using RAxML (v8.2.12)54 with GTRCATX nucleotide substitution model with parameters “-f a -x 12345 -p 12345 -# 500 -m GTRCATX”. With three species divergent time (splits between Gasterosteus aculeatus and Larimichthys crocea, Dicentrarchus labrax and Larimichthys crocea, and Notothenia coriiceps and Gasterosteus aculeatus) from timetree55 used as the calibration time points, we estimated the divergent time between each species by MCMCtree from the PAML package56 with default parameters.
Synteny analysis
The synteny analysis of D. undecimradiatus against L. crocea and T. rubripes was performed on both whole-genome nucleotide level and gene level. On nucleotide level, we used Lastz (v1.02.00)57 to identify synteny blocks with parameters “T = 2 C = 2 H = 2000 Y = 3400 L = 6000 K = 2200”, and aligned blocks with length less than 1 kb were filtered.
On gene level, we used JCVI (v0.8.12)58 to identify synteny genes based on CDS. On JCVI pipeline, sequence alignment was carried out using Lastal (v979) with parameters “-u 0 -P 48 -i3G -f BlastTab”, and then the results were filtered by C-score with parameters “C-score > = 0.70 tandem_Nmax=10”. Finally, we filtered out the syntenic gene blocks spanning less than 5 genes.
Construction of gene trees
To construct a gene tree that truly reflects the evolutionary history, the gene synteny results were used to accurately select orthologous genes. Only genes on 1:1:1 syntenic blocks were extracted and the human gene set was used as an outgroup. For each orthologous gene cluster, we used MUSCLE (v3.8.31)26 to do multiple sequence alignments of CDS with default parameters, and then used RAxML (v8.2.12)54 to construct ML tree using the GTRCATX model with parameters “-f a -x 12345 -p 12345 -# 100”.
Population demographic history inference
The history of effective population size was reconstructed using PSMC (v0.6.5-r67)20. Firstly, diploid genome reference for the individual was constructed using SAMtools and BCFtools59 with parameters “samtools mpileup -C30” and “vcfutils.pl vcf2fq -d 10 -D 100” separately. Then, the demographic history was inferred using PSMC with “−N25 −t15 −r5 −p 4 + 25*2 + 4 + 6” parameters. The estimated generation time (g) and mutation rate per generation per site (μ) were set to 1 and 2.5e−8.
Expansion and contraction of gene families and construction of the phylogenetic tree of gene family
Expansion and contraction of each gene family were identified by Café (v2.1)60 based on the time-calibrated tree. To obtain potential functions of the gene families, the number of different KO terms was counted for each gene family. The functions of the gene families were assigned by the corresponding KO terms with the highest count. The KEGG pathways involved by KO terms were extracted for further functional analysis. To construct phylogenetic trees of gene families, the CDS sequences were extracted to construct maximum likelihood (ML) tree using RAxML (v8.2.12)54.
Source: Ecology - nature.com

