The C. aesculifolia seed batches with positive response to priming present the two main transcriptional phases, as described for Arabidopsis: early and late germination
A principal component analysis (PCA) was performed to evaluate the RWC as a physiological trait during germination (Figs. 4a and 5a). For the PR phenotype, PC1 could explain 61% of the variance between dry and imbibed seeds, and PC2 could explain 26% of the variance between the imbibed stages (16%RWC, 50%RWC, TR and G; Fig. 4a). We obtained differentially expressed genes that show significant changes between different stages and in the overall course of germination, and between physiological stages. The overall expression changes from the dry stage (T0) until radicle protrusion (G) resulted in an up-regulation of 6,635 genes and a down-regulation of 2,457 genes (Supplementary Table S2). The most important changes were detected between T0 and 16% (2,727 up-regulated and 2,375 down-regulated genes); slight changes in gene expression were detected between 16 and 50% (69 up-regulated and 26 down-regulated), but a considerable change between 50% and TR (645 up-regulated and 453 down-regulated) was also detected. A small amount of differentially expressed genes was detected between TR and G (193 up-regulated and 148 down-regulated, Supplementary Table S2). These results suggest that C. aesculifolia seed germination presents the early and late transcriptional phases as described for Arabidopsis13. The GO-term enrichment analyses showed that the biological groups enriched in 16%RWC with respect to T0 were those related to DNA metabolism, transcription and translation, and primary/intermediary metabolism. The response to abscisic acid (ABA), ethylene and cytokinin were also up-regulated. Abiotic stress responses, that included cadmium, cold, heat and salt stress, were upregulated. The response to ABA and abiotic stress were down-regulated. In contrast, only proteolysis related to mobilization of reserve proteins, transmembrane transport and response to water deprivation were enriched at 50%RWC and the down-regulated biological process were related to seed maturation and embryo development (Fig. 4b, Supplementary Table S3).
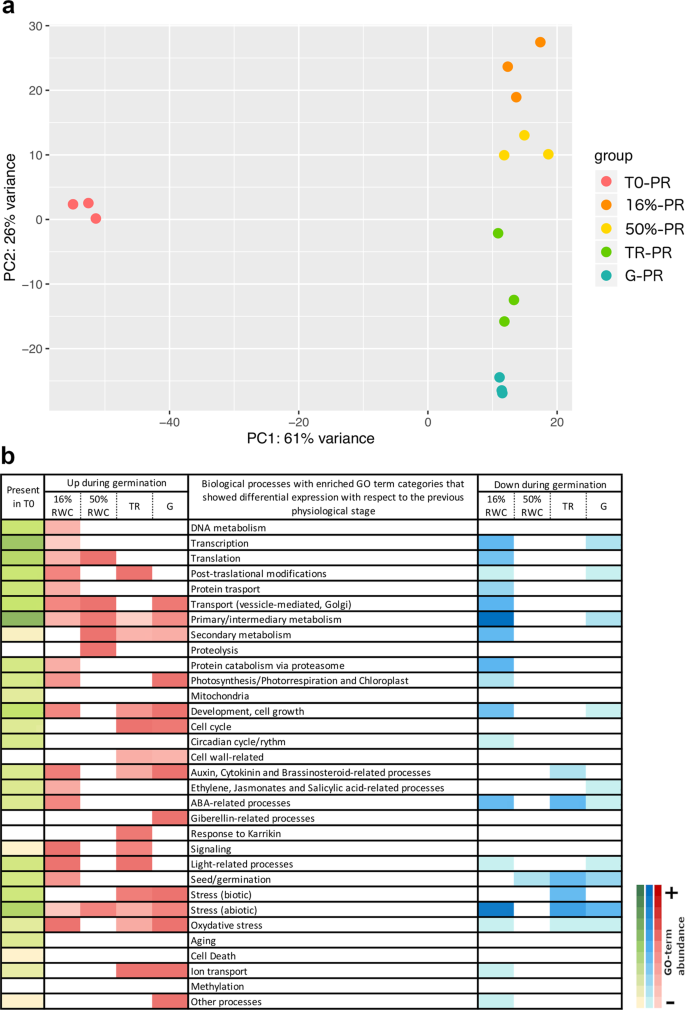
Transcriptome analyses of seed batches with a positive response to priming. In (a) PCA analysis of the transcriptome profiles of three replicates for each physiological stage indicated by colour. PC1 explains 61% of the variation between the dry stage and the imbibed stages. PC2 explains 26% of the variation between imbibed stages. In (b) Main GO-term categories enriched in each physiological stage. The green colour represents the enriched categories present in the dry seed while the red and blue colours represent the enriched categories that had differential expression with respect to the previous stage. Up-regulated categories are shown in red and down-regulated categories in blue.
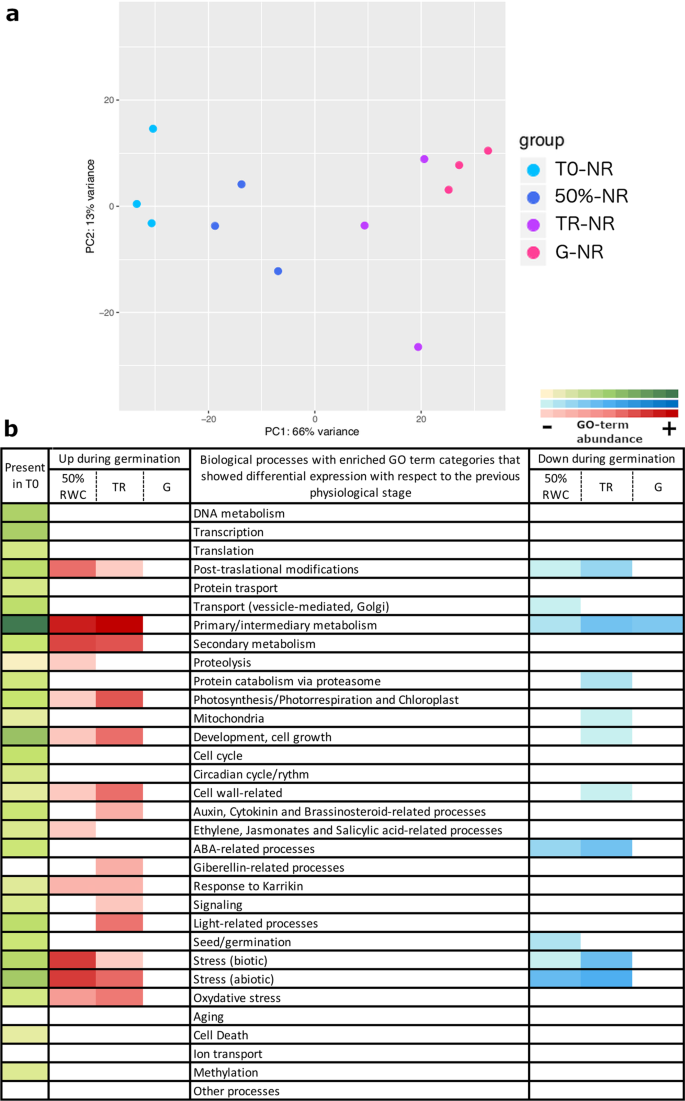
Transcriptome analyses of seed batches with negative/no response to priming. In (a) PCA analysis of the transcriptome profiles of three replicates for each physiological stage indicated by colour. PC1 explains 66% of the variation between the dry stage and the imbibed stages. PC2 explains 13% of the variation between replicates. In (b) Main GO-term categories enriched in each physiological stage. The green colour represents the enriched categories present in the dry seed while the red and blue colours represent the enriched categories that had differential expression with respect to the previous stage. Up-regulated categories are shown in red and down-regulated categories in blue.
The enriched biological processes that represented the TR were primary/intermediary and secondary metabolism, development and cell growth, and cell wall-related processes. The response to cytokinin and auxin were also regulated as well as the response to abiotic and oxidative stresses. The main down regulated category was related to ABA. During G, the main up-regulated categories also included cell wall related processes. The primary metabolism was represented by the up-regulation of lipid metabolism and hormone response included auxin homeostasis and gibberellin response. The carbon fixation and cell cycle processes were strongly up-regulated. Enriched down-regulated biological processes were transcription, lipid storage, sucrose biosynthesis and response to ethylene and auxin.
In the seed batches with negative or no response to priming, the dry seed transcriptome is similar to the imbibed seeds transcriptome in the early germination stages of both phenotypes
For NR phenotype, PC1 could explain 66% of the variation between the different stages (T0, 50%, TR and G) and PC2 could explain 13% between replicates (Fig. 5a). In these seed batches, expression analyses showed moderate changes between stages and in the overall course of germination, in comparison to the observed changes in PR-batches (Supplementary Fig. S5). The overall expression changes from the dry stage (T0) until radicle protrusion (G) showed an up-regulation of 2,437 genes and a down- regulation of 1,856 genes (Supplementary Table S2). In the transition from T0 to 50%RWC a total of 559 up-regulated and 247 down-regulated genes were observed, but a similar amount of expressed genes as the PR-batches between 50%RWC and TR were also detected (762 up-regulated and 470 down-regulated genes). Finally, a small amount of differentially expressed genes was detected between TR and G (21 up-regulated and 56 down-regulated, Supplementary Table S2). The biological groups that were mainly enriched in 50%RWC were those related to primary/intermediary and secondary metabolism (Fig. 5b, Supplementary Table S3). The synthesis of jasmonic acid, and biotic and abiotic stress responses were also upregulated. In contrast to PR-batches, there was not a significant up-regulation of the proteolysis category. Down-regulated biological groups were related to ABA response and embryo development.
The enriched biological processes that represented the TR were primary/intermediary and secondary metabolism, photosynthesis related processes including carbon fixation, development and cell growth and cell wall related processes. The response to light, auxin and gibberellic acid were also up-regulated as well as the response to abiotic and oxidative stresses. The main down-regulated categories were related to ABA and abiotic stress. There were also a down-regulation of biological processes related to post-translational modification and primary metabolism. During G, no significantly enriched up-regulated categories were detected. The enriched down-regulated biological process was related to lipid storage.
The expression and the GO-term enrichment analyses indicated that the germination process in PR-batches occurred accordingly to what has been described in previous studies11,13,26 (Fig. 4b), suggesting that for C. aesculifolia seeds, the transcriptomic profile could be the reference for the proper transitions that arise during germination. However, in the NR-batches, the differential expression and the GO-term enrichment profiles did not reflect, at the same extent, the transitions between physiological stages. We performed time-series analyses and detected eight clusters of gene expression patterns in which the main trends in the PR batches reflected an up- or down-regulation from T0 onwards, while in the NR-batches those same clusters showed subtle or no changes from T0 onwards (Supplementary Fig. S6). These major trends from T0 to 50%RWC suggested that the transcripts levels already present in the dry seeds of the NR-batches were more similar to the observed transcript levels in imbibed seeds. To verify this observation and to test if the RWC could distinguish between physiological stages, despite the underlying differences in the phenotypes, we performed a global PCA and a heatmap with hierarchical clustering analyses (Fig. 6).
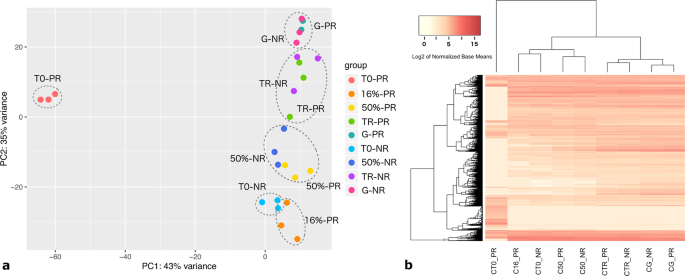
Global analysis of the transcriptome profiles of the two phenotypes during germination. In (a) PCA analysis of the transcriptome profiles of three replicates for each physiological stage indicated by colour. PCA was performed with the top-700 genes that accumulated the most variance across groups. PC1 indicates that the transcriptome profile of the dry stage (T0) in the PR seeds had the most important differences with respect to the other physiological stages, including the dry stage in the NR seeds (43% of variance). PC2 explains 35% of the variance between the remaining physiological stages for both phenotypes. In (b) heatmap of the normalized reads per transcript of all 12,683 Arabidopsis locus tags present in the global transcriptome assembly, and detected in at least one physiological stage. The hierarchical clustering distinguishes the two transcriptional phases13 indicated by low RWCs (16% and 50%) and by TR and G physiological stages. The NR-T0 (dry seed) profile is clustered among the low RWCs, been most similar to 16% transcriptomic profile.
For PR and NR samples, PC1 could explain 43% of the variation between the dry and imbibed seeds except for T0-NR that was grouped with the imbibed seeds, confirming the observation from the clustered expression patterns that the NR-T0 profile is more similar to that of low RWC samples. Meanwhile, PC2 could explain 35% between the different imbibed stages and T0-NR seeds (Fig. 6a). This analysis also demonstrated that the imbibed samples of PR- and NR-batches grouped by their respective physiological stage during the germination process, including 50%RWC, TR, and G. The hierarchical clustering performed with the global transcriptomic data also confirmed that the two transcriptional phases that occur during germination are distinguishable independently of the phenotype as well as the resemblance of the NR-T0 seeds with the low RWCs seeds, by its grouping with the PR-16%RWC samples (Fig. 6b).
In Arabidopsis, the dry seed contains about 12,600 different mRNAs stored during development and maturation27. In the combined transcriptomes of both PR- and NR-dry seeds there was an overlap of about 63% (8,064) of the locus tags reported for Arabidopsis27, and about 37% (3,017) of those were shared between the two phenotypes and Arabidopsis. An incomplete overlap is expected due to the large differences in life histories between the two species. Like Arabidopsis, C. aesculifolia produces endospermic orthodox seeds and presents a two-stage germination, but it is a perennial tree that thrives in warm climates where water availability is the limiting factor for germination to occur. A GO-term analysis of the non-overlapping Arabidopsis locus tags confirmed that almost all those genes belonged to the same gene families within the biological categories presented in Figs. 4 and 5 for C. aesculifolia, especially stress responses, metabolism, and protein transport/modification, thus the differences between the two species is associated with their particular physiology and responses to the environment.
The results confirm that it is possible to compare seed batches of a particular species despite their differences in life histories. Using the transcriptomic profiles of PR-batches during germination as reference, we determined that the NR-dry seeds do present a transcriptomic profile corresponding to the transcriptional phase I of the germination process13. However, they also present some other enriched functional categories that reflect a disarray of the early transcriptional stage, like an overrepresentation of stress-related processes. The premature expression of these processes could reflect a costly and inefficient use of the seed resources, precluding the ability of NR-batches to properly respond to the priming treatment or other external stimuli.
The NR-batches are capable of adjusting their transcriptomic profiles during the germination process, despite their initial differences with respect to the PR-batches
The overlap of the six samples at the moment of radicle protrusion (G), and the clustering of samples by physiological stages suggested that the transcriptomic profiles of PR and NR-batches gradually become more similar during germination (Fig. 6). This is also observable in the clusters in Supplementary Fig. S6, since none of the eight clusters showed a trend in which all the genes from that particular cluster had a reversed expression trend between PR- and NR-batches (i.e. that the PR-batches had a trend of up-regulation at G while the NR-batches displayed a down-regulation trend at G). This trend of PR- and NR-batches to converge in transcriptomic profiles by the end of the germination process was observed even in the simulations run with a higher number of clusters or cluster membership stringency. The differential expression analyses between PR- and NR-batches for each physiological stage confirmed a reduction in the total number of differentially expressed genes during late germination (Fig. 7).
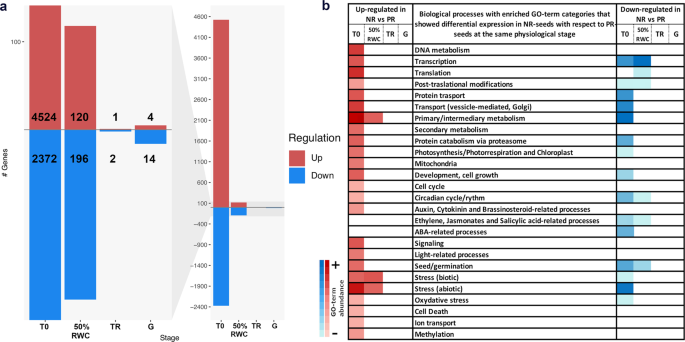
Differentially expressed genes in NR-batches with respect to PR-batches in each physiological stage. In (a) total gene count of differentially expressed genes in each physiological stage from the dry seed (T0) to the moment of radicle protrusion (G); the y axis represents total gene count. The left panel corresponds to a zoomed image of the graph in the right panel. In (b) GO-term enrichment in each category of biological processes represented in the gene set. The darker colours in the blue or red scales represents abundance of GO-terms and gene count within each category. The physiological stages TR and G had no particular enrichment of any category due to the small amount of genes detected.
For T0 79% of the present transcripts showed differential expression, while only a 4%, 0.03% and 0.2% of transcripts present in 50%RWC, TR and G respectively were affected. In the NR-T0 seeds it was detected a GO-term enrichment in 24 of the 32 categories of biological processes in Figs. 4 and 5, of which primary/intermediary metabolism, followed by abiotic stress, translation and, DNA metabolism were the categories which presented the most abundance of terms and up-regulated genes (Fig. 7b; Supplementary Table S3). In the case of the down-regulated genes, the main categories were again the primary/intermediary metabolism as well as abiotic stress, which presented the most abundance of GO-terms and genes; but also vesicle transport, protein transport and transcription showed an important enrichment. At the 50%RWC stage, primary/intermediary metabolism, biotic and abiotic stresses were the categories that presented GO-term enrichment, while transcription was the main down-regulated category, followed by seed/germination-related processes, translation, post-translational modifications, circadian rhythm, and ethylene/jasmonic acid-related processes. For TR and G stages, no GO-term enrichment was detected. This result suggests that, although the transcriptional profile of NR-T0 is more similar to the early germination profile, these seeds are capable of adjusting their transcript levels during imbibition in order to maintain the germination program.
Finally, transcript profiles and differential expression between PR- and NR-batches and during the germination process was verified by RT-PCR in five randomly selected genes from the data set (Fig. 8). The data was normalized using ACT7 (At5g09810) as reference28. Two of these selected genes correspond to a large gene family associated to primary cell wall modification (PME3, At3g14310, Fig. 8a,f; an inhibitor of PME activity At5g20740, Fig. 8b,g), and have been described in germination as playing important roles in testa and endosperm rupture during germination in many species29,30,31. In accordance with other studies, our RNAseq data and the RT-PCR verification showed that these two genes were consistently up-regulated in both PR- and NR- batches during germination, being at the moment of TR and G where the signal was more prominent. The gene GDPD1 (Fig. 8e,j) is associated to phospholipid metabolism, and represents the down-regulation trend in the overall gene expression during germination. No differential expression was detected between PR- and NR- batches at any stage in the RNAseq data set, and this was verified by the PCR assay. This gene has been reported in Arabidopsis to be down-regulated during germination, with transcript levels being undetected as soon as 3 hours after imbibition32,33. In C. aesculifolia, a similar trend was detected, although the gene is still detectable by the end of germination. The two genes that represent the differential expression between PR- and NR- batches correspond to HDH (At4g04320, Fig. 8c,h), involved in degradation of valine for energy production34, and XERICO (At2g04240, Fig. 8d,i), a master regulator of ABA metabolism and participates in osmotic and drought stress responses in seedlings and adult plants35. These two genes are up-regulated in T0 of NR-batches with respect to PR-batches, but their expression profiles converge by the end of germination. Overall, these genes reflect the main trends in expression changes detected in the data set and confirm that despite the initial differences in the dry seed between the two phenotypes, the germination program occurs in an orderly and stage-specific manner.
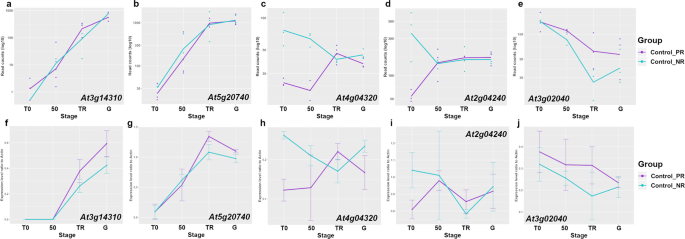
RNAseq expression profiles (a–e), and semi-quantitative RT-PCR validation (f–j) of the differential expression of five selected genes. RT-PCR data is represented as the ratio with respect to ACT7 (At5g09810) expression. Purple lines depict the average read counts (a–e) and average ratios (f–j) of the seed batches with positive response to priming (PR), while the turquoise lines depict the seed batches with no response/negative response to priming (NR). The dots in the upper row of panels depict the normalized read counts for each library, while the vertical bars in the lower row panels represent standard deviation of three independent PCR replicates.
Ecophysiological implications of the changes in the transcriptomic profiles of dry seeds
Even in a metabolic quiescent state such as the mature dry seed, there are a series of changes that occur at low water contents affecting diverse cellular components and macromolecules36,37,38. Seed after ripening is a process that occurs in the dry stage that can widen or increase the seeds sensitivity and perception of environmental conditions promoting germination39, and has been demonstrated to be a discrete developmental process40 that allows for reversible changes in the overall transcriptomic and proteomic profiles. This allows the embryo to adjust in response to environmental stimuli before the commitment to initiate the germination program41. From an ecological perspective, seed release from the mother plant in the adequate season allows for the after ripening and natural priming to occur and proper synchronization of germination with the rainy season. The PR-batches displayed a phenotype that could readily incorporate the external signal produced by the priming treatment by allowing a gradual advancement of the germination program as a fine-tuning strategy to reduce the seed-batch average time needed to complete germination once the water stimulus became stable.
In the NR-batches there are two different life histories, associated with time in storage, that converge in a loss of the capability to respond to the priming treatment and resemble an “imbibed” seed. In the 2016 seed-batch, the failure to response to priming could be a result of stressful environmental conditions experienced by the mother plants, which generated an untimely release of seeds, and a change in the transcriptome that disrupted the proper transition from seed development/maturation to the quiescent stage in which after ripening occurs38,41. However, for the stored batches, the seeds mRNA degrades continuously during storage37,38, and in a recent study, it was demonstrated that those changes can be detected even before notably changes in germination performance are perceived by standard longevity and vigour tests. These changes in the transcriptomic profile are especially notorious in longer transcripts, which can degrade faster than shorter transcripts42. This finding is in accordance with our rationale of studying the shifts in the transcriptome by analysing only those assembled sequences that have the features of a complete coding sequence, since the total read count would reflect only those sequences with the highest probability of still being fully functional in the seeds context. Thus, the germination capacity of the stored NR-batches in our tested conditions, along with their respective transcriptomic profiles, indicate that these seed batches might have some extent of the deleterious effects associated to storage24,37,38, but the transcript profiles points to a scenario of an extended and disrupted after-ripening process rather than a deleterious effect on the germination program. Overall, the advancement of the transcriptomic profile in the NR-T0 seeds could reflect a risky gamble for survival (“bet-hedging”)43, by decoupling the germination program from the hydric signal in order to favour germination in a wider range of conditions, but the risk of survival could be re-assigned to the next developmental phase and have cascading effects on fitness.
Phenotypic variability within and among individuals of a particular species or group of species could be a positive feature for ensuring organismal fitness and can be studied at different levels44. Phenology transitions are varying as climate shifts, so these changes in the timing of developmental events, important for determining organismal fitness45, pose another challenge to the comprehension of the basic molecular and physiological processes driving the interaction between organisms and their environment. We demonstrated that the use of physiological traits, specific to a particular developmental stage, is a reliable time-independent approach to detect common patterns between groups with wide variability and plasticity.
Source: Ecology - nature.com
