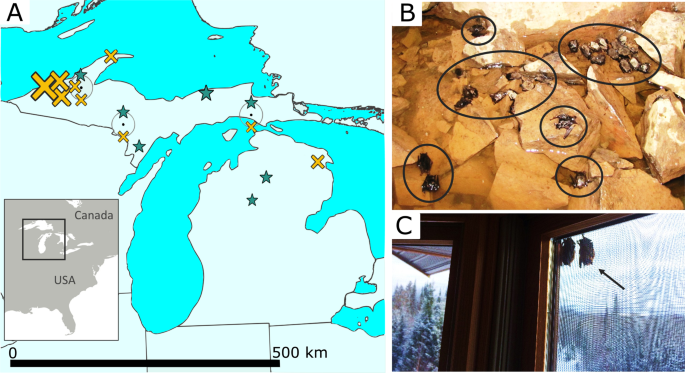
Study area
We chose northern Michigan, USA, for our study because it represents a reasonably isolated population of little brown bats (Fig. 1A); WNS is present throughout our study area, and was first detected there in early 2014. We sample non-survivors from hibernation sites during the winter and survivors during the summer (when they are no longer afflicted by the pathogen). However, because the species utilizes short distance seasonal migration (typically ≤ 500 km64), during warmer periods they do not roost in the same sites in which they hibernate, thus the relative geographic isolation is important for assuring that bats sampled during both seasons were from the same population. Winter hibernation sites are concentrated in the northwestern portion of our study area (hibernation sites are lacking in the central and southern Michigan), and primarily consist of abandoned iron and copper mines. As a consequence, bats in our area (Fig. 1A) are isolated from other populations by two factors: the Laurentian Great Lakes and the lack of suitable subterranean hibernation sites within migration range in central and southern Michigan. The seasonal sampling of bats is necessary because WNS non-survivors can only be documented in winter areas, and disease survivors can only be identified during summer.
Sampling of focal species
All sampled bats (Table S1) were categorized as either “survivors” or “non-survivors” of WNS. Survivors (n = 9) were adult bats that had been born the previous year or earlier and thus had survived at least one hibernation period with the WNS pathogen (collected during summer s of 2016–2017, see Anthony65 for aging methodology). Most individuals which succumb to the disease are found within the subterranean sites that afflicted species of bats rely upon in winter, and in which the fungus thrives, however some infected bats leave hibernation sites prematurely in winter in search of food or water, but quickly die due to lack of available resources and sub-freezing temperatures. Correspondingly, most non-survivors we sampled were bats found dead in or near hibernation sites during winter (collected in early 2016; n = 25; Fig. 1), although some tissue samples came from individuals with the pathogen that were euthanized during surveillance studies (i.e., they tested positive for the fungus; collected in early 2014; n = 4). Note that comparing survivors to this more general group of non-survivors makes tests for loci under selection more conservative, in that some of the euthanized bats categorized as non-survivors may not have died from WNS naturally. However, if non-survivors actually carried adaptive alleles, this would not produce a bias (i.e., make it more likely) to detect putatively selected alleles—in fact it would make such detection more difficult. In addition, all analyses were repeated excluding the euthanized bats to confirm the robustness the results.
Samples for most non-survivors (n = 23) were from bat carcasses found during winter either in or proximal to the caves or mines in which they were hibernating. Prior to the introduction of WNS, it was uncommon to find dead bats at hibernacula, whereas conspicuous numbers of dead individuals are found in and around these sites post-introduction of the disease (Fig. 1B), and all sites were WNS-positive at the time of collection. The accidental inclusion of bats which had died due to other causes would make it more difficult to detect adaptation in our analyses. To reduce disturbance to hibernating bats, dead bats were collected in conjunction with routine surveys by the Michigan Department of Natural Resources (MDNR) and Eastern Michigan University. Four samples were contributed by the U.S. Geological Survey National Wildlife Health Center; these bats were found during hibernation with the fungus growing on them, but were euthanized (as discussed above). Lastly, two samples of non-survivors came from the MDNR Wildlife Disease Laboratory (see details below).
Among the survivors, collection methods varied (Table S1). Three survivors were captured during summer using mist-nets, and visual inspection confirmed evidence of recovering from WNS (i.e., the presence of healing wing lesions or scars). Tissue samples were collected via small biopsy punches (2 mm diameter, one punch for each wing, Premier Medical Products Company, Plymouth Meeting, Pennsylvania, USA), after which bats were immediately released. No individual was detained for longer than 30 minutes. Eight specimens were contributed by the MDNR Wildlife Disease Laboratory, which annually receives large numbers of bats for rabies testing after they are encountered by humans or pets66. All individuals used in this study tested negative for the rabies virus. Six of these were considered survivors because they were submitted for testing in summer or fall; during the summer this species uses structures such as houses in addition to trees40 so there is no reason to believe that animals encountered by people during warmer periods were unhealthy. However, at the latitude of our study, little brown bats are not known to hibernate in buildings40. Consequently, any individual encountered by humans during sub-freezing periods is almost certainly on the cusp of dying from WNS. Individuals submitted to the MDNR Wildlife Disease laboratory in winter or early spring were therefore assigned to the non-survivor group (n = 2 in this study).
DNA sequencing and data processing
DNA was extracted from membrane of wing tissue using DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA, USA) and used to prepare a reduced representation genomic library for sequencing. Two restriction enzymes, EcoRI and MseI, were used to digest extracted DNA (ddRadSeq33), to which barcodes (unique tags 10 base-pairs long) and adapters for Illumina sequencing were then ligated. Ligation and amplification were done via polymerase chain reaction (PCR), and 350 to 450 bp long fragments were size selected using Pippin Prep (Sage Science, Beverly, Massachusetts, USA). The library of 38 samples was sequenced in one HiSeq. 2500 lane (Illumina, San Diego, CA, USA), at the Centre for Applied Genomics (Toronto, Ontario, CA).
Genomic sequences were demultiplexed using the STACKS bioinformatics pipeline67 (v. 2.1; specifically process rad-tags, gstacks, and populations), and processed in conjunction with supporting programs. The first step, process radtags, allowed up to one mismatch in the adapter sequence and two mismatches in the barcode, with rescue of RAD-Tags allowed. A sliding window of 15% of the read length was used for an initial exclusion of any reads with a Phred score68 below 10 within the window (note additional filters of a minimum Phred score of 30 were applied in downstream processing, as discussed below). Of 102,419,857 initial sequences, process radtags removed 1,144,865 reads containing the adapter sequence, 18,775,218 reads with ambiguous barcodes, 156,274 low quality reads, and 2,495,192 reads with ambiguous RAD-Tags.
We then indexed a previously generated reference genome for the species, ftp://ftp.ncbi.nih.gov/genomes/Myotis_lucifugus (7x coverage; MYOLUC v. 2.041), and mapped our sequences to the genome using the Burrows-Wheeler Alignment Program (v. 7.17) indexing and MEM algorithms, respectively69,70. The resulting files were filtered (-F 0x804, -q 10, -m 100), converted to .bam files, and sorted using SAMtools71,72 (v. 1.8-27).
The reference-based method of gstacks (set to remove PCR duplicates) was run using the Marukilow model73, minimum Phred68 score of 30, and alpha thresholds (for mean and variance) of 0.05 for discovering single nucleotide polymorphisms (SNPs). This resulted in 59,888,201 BAM records and 581,607 loci (8% of reads were excluded because they were excessively soft-clipped, and 3% had insufficient mapping qualities to be included). All remaining loci were genotyped, with a mean per-sample coverage of 10.5x ± 7.1x, a mean of 138.5 bps per locus, and consistent phasing for 88.3% of diploid loci.
Populations was then run with default settings and the resulting loci were filtered with a custom script in R74 (v. 3.5.0) to remove loci and SNPs that may be artifacts of sequencing or alignment errors (Fig. S5) based on the number of SNPs per read position, resulting in exclusion of SNPs occurring in the last 2 bp of each read. Loci with unusually high levels of diversity were also removed from consideration (threshold θ > 0.026), leaving 273,261 unique loci.
Using the list of vetted loci and SNPs, populations was then run again, retaining loci present in at least 56% of both survivors and non-survivors, ensuring a minimum sample size of at least six survivors; note the actual missing data was typically much lower (i.e., <15% in all but 7 individuals of survivors and non-survivors). This resulted in 40,963 loci (140-bp segments), of which were variable, containing 19,797 SNPs (our final SNPs), all of which had a minor allele frequency of >0.01. Minor allele thresholds of 0.01 and 0.05 were evaluated for downstream analyses, and when warranted the higher threshold was used (noted below). Mean genotyped sites per locus was 142.41 bp (SE ± 0.02). Because some loci contained more than one SNP, the robustness of downstream analysis to inclusion of multiple versus a single SNP per 140-bp fragment was evaluated. Main findings did not differ, thus we present analyses based on multiple SNPs per locus in the main text (see Fig. S6 for results based on a single SNP per locus).
We also checked that the data were not biased due to different levels of genetic decomposition between the survivors and non-survivors by analyzing the Guanine-Cytosine (GC) content of each sample. Specifically, raw Illumina reads (immediately after process radtags) of survivors were compared with the non-survivors using BBMap75 (v. 38.01). The proportion of GC per individual per locus was averaged across all loci for each individual using a custom script in R. Mean GC content was 43% for survivors (n = 9) and 42% (n = 29) for non-survivors, which confirmed non-survivors were not biased towards higher GC because of decomposition.
In addition, the relatedness of sampled individuals was evaluated in two ways: with related76 in R74 and using Plink77. Due to program constraints in related, 250 loci were randomly selected to simulate 100 pairs of individuals in each of four categories: parent-offspring, full sibling, half sibling, and unrelated. Application of the Ritland estimator of relatedness78 to both the simulated and empirical dataset of 1,242 filtered SNPs (see Fig. S7 caption) indicated that none of the individuals in our dataset were related with the exception of two of the non-survivors, which may be half-siblings (Fig. S7). However, the Plink77 analysis of 6,237 SNPs (restricted to a single SNP per locus and minor allele frequency >0.05, as per guidelines) indicated no related individuals within our dataset. We kept all individuals in downstream analyses, because the presence of a single pair of potential half siblings is not expected to influence estimates of allele frequencies or FST, and removal of putatively related individuals can actually increase the error (for more details, see Waples & Anderson79).
Lastly, to confirm that individuals from different sampling sites within the study area could be considered one population, we used Structure37(v. 2.3.4) to evaluate if genome-wide differentiation indicated a single, panmictic population. We selected the ADMIXTURE model with ‘Allele frequencies correlated’ turned on and no prior information about sampling population and explored the best supported model, considering a range of genetic clusters (i.e., k = 1 to 5) with 10 repetitions for each k, for 500,000 Markov chain Monte Carlo iterations with a burn in of 50,000. Visual assessment was used to ascertain convergence by examining plots of FST, alpha, and likelihood versus iterations, and to check for consistency among the ten iterations. No evidence of genetic subdivision based on geographic sampling locality was detected (see Fig. S8).
Tests of genetic drift
Given the large numbers that have died from WNS in this species, genetic differentiation between survivors and non-survivors may result because some alleles, just by chance, will increase or decrease in frequency. These stochastic, non-adaptive genomic changes in otherwise neutral portions of the genome (genetic drift) can be particularly great when only a small proportion of the population survives, sometimes causing population bottlenecks. To visualize the drift-induced changes that occurred broadly across the genome, we conducted a principal components analysis (PCA) of the survivors, and projected the non-survivors onto the estimated PC axes, and the degree of drift was quantified using the F-model36 in STRUCTURE37.
The PCA was calculated for the survivors, onto which the non-survivors were projected (by applying the same scaling and centering used for survivors to the non-survivors; see Lipson et al.80). Generating a PCA in this manner is a method of visualizing differences when one group is a subset of the other (in terms of the proportion of variance), for example due to a series of founder events80.The PCA was performed in R74, in conjunction with the packages Adegenet81 (v. 2.1.1) and Plyr82 (v. 1.8.4) using the prcomp function. One survivor and four non-survivors were excluded from this analysis because of missing data (i.e., >50% missing loci), as were loci missing in >50% individuals (data were filtered using Plink v. 1.0777; see Table S1). After this, the actual missing data was <15% for all individuals except one survivor and one non-survivor, with just under 50% missing data. Missing data were then replaced with the per locus mean value across all individuals. Only genomic sites with a minor allele frequency of ≥0.05 that were variable in both survivors and non-survivors were considered, for a total of 11,462 SNPs. The PCA was repeated to confirm the robustness of the results to missing data threshold, this time using a minimum data threshold of 8.7% missing data per individual and 19% per locus (mean missing data was 1.9%), which resulted in 13,666 loci and 31 individuals being included.
We also directly estimated the amount of genetic drift between survivors and non-survivors in Structure37 using the F-model36 (see also Harter et al.83.). The F-model accounts for differences in population sizes, and has been used to quantify differences in drift between groups of contrasting sample sizes that are similar in proportion to our own83. For our parameter of interest, F, we used a prior mean and SD of 0.10, which places similar probabilities on both large and small values of F. To implement this Bayesian approach, we preassigned individuals to one of the two groups (survivor or non-survivor), and used a burn-in of 50,000 followed by 500,000 reps. We fixed lambda at 1, and used a uniform prior from 1 to 10 for alpha, with a standard deviation of 0.025. Three iterations were run, with different random seeds for initiating the Markov Chains.
Tests of loci under selection
To identify genetic differences among individuals that might have contributed to their survival of WNS, we used FST-outlier analyses, where the signature of selection can be detected by considering the proportional split of allelic variants between groups relative to background levels across the genome34,35. We identified candidate loci using three methods of outlier detection—identification of outliers via (i) the number of standard deviations from the mean using an AMOVA-corrected FST84, (ii) by assessing confidence intervals from bootstrap permutation across loci, and (iii) measuring departure from a chi-squared distribution (detailed below). Variable sites which met all three requirements were regarded as candidate loci apparently undergoing positive selection. All tests of selection were conducted with and without the four non-survivors sampled in 2014 (collected prior to the other specimens), to confirm that the results were robust. Note that the low number of sampled survivors reflects the devastating impact of WNS on this species; despite the small sample size, it is not beyond a size in which SNPs under selection can be detected with FST-outlier analyses85.
In our first approach, we used the AMOVA-corrected FST84 calculated by populations in STACKS35. SNPs with an FST-value of greater than nine standard deviations from the mean (mean = 0.018 ± 1 SD of 0.026) were considered outliers (similar to Willoughby et al.42). A threshold of five standard deviations is often used in detection of outlier SNPs under positive selection42,86,87. We increased our threshold of significance to nine standard deviations to reduce the potential for false-positives. In the second approach, confidence intervals (95% CI) were estimated using diveRsity88. Using the diffCalc function, Weir and Cockerham’s FST89 was calculated for all loci, with 1,000 bootstraps performed across loci. Only loci for which the lower limit of the CI remained five SD from the mean were considered outliers. In the third approach, outliers were identified with OutFLANK90, which estimates the expected neutral variation of FST-values under a chi-squared distribution. As per the developer guidelines90, we excluded loci with low expected heterozygosity (<0.1), and visually adjusted the trim functions to best fit the observed distribution (LeftTrimFraction = 0.3 and RightTrimFraction = 0.05; Fig. S9). Significance was assessed using qvalue91 in R74 (v. 2.12).
All results were visualized in R74, often in conjunction with the package ggplot292. A custom script was used to identify SNPs which were identified as candidate loci under all three methods, and putatively selected sites were then cross-referenced with the species’ annotated reference genome41 to infer possible phenotypic function (see93,94 for additional information on the reference genome and annotation). If the SNP’s position was not within a gene, the nearest annotated areas in each direction were identified.
Source: Ecology - nature.com
